
Abstract
Background
Atogepant is an oral calcitonin gene–related peptide receptor antagonist approved in the US and EU for the preventive treatment of migraine in adults. We evaluated the efficacy, safety, and tolerability of atogepant for the preventive treatment of episodic migraine (EM) in Japanese participants.
Methods
RELEASE was a phase 2/3, multicenter, randomized, double-blind, placebo-controlled study enrolling adult participants with a ≥1-year history of migraine, <50 years of age at time of migraine onset, history of 4–14 monthly migraine days (MMDs), and <15 monthly headache days in the three months prior to screening and during the screening/baseline period. The study included a four-week screening/baseline period, 12-week double-blind treatment period (DBTP), 12-week active treatment extension period, and 30-day safety follow-up. Participants were randomized 1:1:1:1 to placebo, atogepant 10 mg once daily (QD), 30 mg QD, or 60 mg QD for the 12-week DBTP. Completers of the DBTP could continue to the 12-week active treatment extension period where the placebo group was rerandomized 1:1:1 to atogepant 10 mg, 30 mg, or 60 mg; atogepant groups continued the same dose. The primary endpoint was the change from baseline in mean MMDs across the 12-week DBTP.
Results
Of 807 participants screened, 523 were treated in the 12-week DBTP (Safety Population 1 [placebo, N = 134; atogepant 10 mg, N = 126; 30 mg, N = 131; 60 mg, N = 132]; modified intent-to-treat population [placebo, N = 133; atogepant 10 mg, N = 127; 30 mg, N = 130; 60 mg, N = 131]). The least square mean difference (95% confidence interval) from placebo in mean MMDs across 12 weeks was −1.57 (−2.24, −0.89) for atogepant 10 mg, −1.90 (−2.57, −1.22) for 30 mg, and −2.10 (−2.78, −1.43) for 60 mg (all p < 0.0001). Treatment-emergent adverse events (TEAEs) in the DBTP occurred in 46.3%, 45.2%, 38.9%, and 43.2% of participants receiving placebo, atogepant 10 mg, 30 mg and 60 mg, respectively. During the DBTP, TEAEs occurring ≥5% were constipation and nasopharyngitis, and there was one serious TEAE in the atogepant 10 mg group considered not related to treatment. TEAEs resulting in treatment discontinuation were infrequent in all treatment groups in the DBTP. Safety was consistent in the 12-week active treatment extension period.
Conclusions
Atogepant treatment demonstrated statistically significant and clinically meaningful reductions in mean MMDs compared with placebo across the 12-week DBTP in Japanese participants with EM. The safety profile of atogepant in Japanese participants was consistent with the known safety profile in the global population. No new safety signals were identified.
Trial registration
ClinicalTrials.gov NCT05861427; https://clinicaltrials.gov/study/NCT05861427
This is a visual representation of the abstract.
Introduction
Migraine is a neurological disease characterized by recurring attacks of headache accompanied by sensitivity to light and sound, nausea, and vomiting (1,2). Migraine impacts an estimated 1.1 billion individuals worldwide, and the prevalence of migraine in Japan has been reported to be 6.0%–8.4% (3–5). Migraine is the second leading cause of disability globally, and the leading cause of disability in young women (6,7). Of those with migraine, >90% have episodic migraine (EM), which is characterized by <15 headache days per month (8–10). The management of migraine includes acute treatments that relieve pain and disability after the onset of the headache and preventive treatments, which prevent the attack from occurring (1).
The Japanese Society of Neurology clinical treatment guidelines state that preventive treatments for migraine should be considered for individuals who experience migraine more than twice a month, have significantly impaired quality of life or daily functioning, have an insufficient response or contraindication to acute treatments, or have a risk of medication overuse headache due to frequent use of acute treatments (11). A retrospective analysis of Japanese health insurance claims data found that preventive treatment use was low (5.4% receiving only preventive treatment, 9.5% receiving acute and preventive treatment), discontinuation rates were high, and only 15% of patients restarted treatment after discontinuation, showing a high proportion of Japanese patients remaining untreated with preventive treatment (12). Additionally, Japanese physicians have fewer preventive treatment options for migraine compared to the United States (13). Together, there remains a high unmet need in the Japanese population for newer migraine preventive treatments that are safe and well tolerated with demonstrated efficacy in EM.
Calcitonin gene-related peptide (CGRP) is a neuropeptide with an established role as a key mediator of migraine attacks (14,15). Treatments targeting CGRP or its receptor have been developed for the preventive treatment of migraine, and these include monoclonal antibodies (mAbs) and gepants, or oral CGRP receptor antagonists. Atogepant is an oral CGRP receptor antagonist currently approved in the US and EU for the preventive treatment of migraine in adults (16,17). The efficacy and safety of atogepant for the preventive treatment of EM has previously been reported in one phase 2b/3 (NCT02848326) and two phase 3, randomized, double-blind, placebo-controlled, 12-week trials (ADVANCE [NCT03777059], ELEVATE [NCT04740827]) (18–20). These trials consisted of a patient population of predominantly Caucasian individuals. Here we present the first evaluation of the efficacy, safety, and tolerability of atogepant for the preventive treatment of EM exclusively in a Japanese population.
Methods
Study design
RELEASE (NCT05861427) was a phase 2/3, multicenter, randomized, double-blind, placebo-controlled, parallel group study conducted at 48 sites across Japan (Online Supplemental Table 1). The study consisted of a 4-week screening/baseline period, 12-week double-blind treatment period, 12-week active treatment extension period, and a safety follow-up period 30 days after the last dose of study treatment (Online Supplemental Figure 1). Participants were enrolled from August 2023 to August 2024 and those who completed the four-week screening/baseline period and met all entry criteria were randomized to the double-blind treatment period of the study at Visit 2. Participants were randomized 1:1:1:1 using interactive web response technology to placebo, atogepant 10 mg once daily (QD), atogepant 30 mg QD, or atogepant 60 mg QD. Randomization was stratified by prior exposure (yes/no) to a migraine preventive treatment with proven efficacy and by site. The study enrolled approximately 70% of randomized participants with ≥1 prior migraine preventive treatment with proven efficacy. Visits occurred every four weeks during the double-bind treatment period until Week 12 (Visit 5).
Completers of the double-blind treatment period were allowed to continue into the active treatment extension period. Participants who were randomized to the atogepant treatment groups in the double-blind treatment period continued to be assigned to the same dose treatment group in the active treatment extension period. Participants in the placebo group in the double-blind treatment period were rerandomized to atogepant 10 mg, atogepant 30 mg, or atogepant 60 mg treatment groups (dose blinded) in a 1:1:1 ratio to enter the active treatment extension period. Visits occurred every four weeks in the active treatment extension period until Week 24 (Visit 8).
Participants
The study enrolled adult participants with ≥1-year history of migraine, defined using the International Classification of Headache Disorders, 3rd edition (ICHD-3) (2) criteria, <50 years of age at the time of migraine onset, history of 4–14 migraine days and <15 headache days per month in the three months prior to screening and in the 28-day baseline period, and completed ≥20 out of 28 days of the eDiary at the baseline period.
Participants were excluded if they had difficulty distinguishing migraine headaches from tension-type or other headaches, a history of migraine accompanied by diplopia or decreased level of consciousness, or retinal migraine as per ICHD-3, or a current diagnosis of chronic migraine, new daily persistent headache, trigeminal autonomic cephalalgia, or painful cranial neuropathy. Participants with ≥15 headaches per month on average across the three months prior to Visit 1 or in the 28-day baseline period were excluded. Participants with any other concurrent pain condition that, in the investigator's opinion, may significantly impact the current headache disorder (e.g., fibromyalgia, facial pain) were excluded.
Participants who had an electrocardiogram (ECG) with clinically significant abnormalities at screening, clinically significant cardiovascular or cerebrovascular disease per the investigator's opinion, or hypertension (sitting systolic blood pressure >160 mm Hg and/or sitting diastolic blood pressure >100 mm Hg) were excluded. Additionally, participants were excluded if they had clinically significant hematologic, endocrine, cardiovascular, pulmonary, renal, hepatic, gastrointestinal, or neurologic disease (unless stable for >1 year by investigator judgement). Participants were excluded if they had confounding psychiatric conditions, dementia, epilepsy or significant neurological disorders other than migraine, in the opinion of the investigator, or any clinically significant laboratory values. Participants that were pregnant, planning on becoming pregnant, or currently lactating were excluded.
The following medications for the acute treatment of migraine were allowed during the study: triptans, ergot derivatives, ditans, opioids, other forms of analgesics (including acetaminophen), nonsteroidal anti-inflammatory drugs (NSAIDs), and antiemetic agents. Those who had used opioids or barbiturates >2 days/month, triptans, ergots, or ditans ≥10 days/month, or simple analgesics (e.g., aspirin, NSAIDs, acetaminophen) ≥15 days/month in the three months prior to Visit 1 per investigator's judgment or during the baseline period were excluded from the study. Additionally, participants with a history of an inadequate response to >4 medications (two of which have different mechanisms of action) prescribed for the preventive treatment of migraine were excluded. Medications with demonstrated efficacy for the preventive treatment of migraine (e.g., amitriptyline, topiramate, propranolol) were prohibited 30 days prior to Visit 1 and throughout the study. The use of an injectable mAb blocking the CGRP pathway or therapeutic or cosmetic botulinum toxin injections into the head, face, or neck in the previous six months, or more than three doses of ubrogepant, rimegepant, or zavegepant were prohibited. Participants with previous exposure to atogepant were excluded.
Outcomes
The primary outcome of RELEASE was the change from baseline in mean monthly migraine days (MMDs) across the 12-week double-blind treatment period. Baseline was defined as the number of migraine days during the last 28 days prior to randomization. Secondary efficacy endpoints included the change from baseline in mean monthly headache days (MHDs), mean monthly acute medication use days, and proportion of participants achieving a ≥50% reduction in mean MMDs across the 12-week double-blind treatment period. Mean change from baseline in MMDs at Weeks 1–4, 5–8, and 9–12 was a prespecified additional analysis. An eDiary was used to record the daily total duration of headache, headache characteristics, associated symptoms (e.g., nausea/vomiting, photophobia or phonophobia, and aura), the worst pain severity, and acute medication use both in the screening/baseline period and double-blind treatment period until the end of the double-blind treatment period. Training for the eDiary was provided for qualified participants during the screening visit. A migraine day, headache day, and acute medication use day are defined in the Online Supplemental Methods.
Secondary endpoints for patient-reported outcomes (PRO) on quality of life and ability to function included change from baseline in mean Migraine-Specific Quality of Life Questionnaire v2.1 (MSQ v2.1) Role Function-Restrictive (RFR) domain score at Week 12, and mean Activity Impairment in Migraine-Diary (AIM-D) Performance of Daily Activities (PDA) and Physical Impairment (PI) domain scores across the 12-week double-blind treatment period. MSQ v2.1 is a 14-item, 3 domain questionnaire that measures health-related quality of life impairments attributed to migraine in the past four weeks. The RFR domain assesses how migraine impairs an individual's daily social and work-related activities and was prioritized for evaluation as a secondary endpoint in this study, as well as other atogepant studies (18,19), as it assesses proximal migraine impacts on daily activities meaningful to patients with EM. AIM-D is an 11-item, 2 domain PRO measure that assesses the level of difficulty individuals have with performing daily activities (PDA domain) and the level of physical impairment (PI domain). MSQ v2.1 was collected on an eTablet at specific clinic visits and AIM-D was collected via the daily eDiary. Additional endpoints include the change from baseline in MSQ v2.1 RFR domain score at Weeks 4 and 8, Role Function-Preventive (RFP) and Emotional Function (EF) domain scores at Weeks 4, 8, and 12, and AIM-D total score, PDA domain score, and PI domain score at Weeks 1–4, 5–8, and 9–12. The established between-group minimally important difference (MID) for the MSQ v2.1 RFR, RFP, and EF domains are ≥3.2, ≥4.6, and ≥7.5, respectively (21). AIM-D has previously been psychometrically validated in a 13-week prospective, observational study and using phase 3 clinical trial data (22–24). Detailed methods for MSQ v2.1 and AIM-D have been previously published (25).
Safety evaluations included the incidence of adverse events (AEs), serious AEs, AEs leading to discontinuation, and vital sign measurements, ECG variables, clinical laboratory evaluations (hematology, chemistry, urinalysis), and Columbia Suicide Severity Rating Scale (C-SSRS) for the entire study duration. Treatment-emergent AEs (TEAEs) were defined as AEs with a recorded onset date on or after the date of the first dose of study treatment in the respective treatment period until 30 days after the last dose of study treatment. An AE is considered a TEAE for the double-blind treatment period if the onset date is after the first dose of study treatment and ≤30 days after the last dose of double-blind study treatment or the day before the date of the first dose of the active treatment extension study medication, whichever is earlier. An AE is considered a TEAE for the active treatment extension period if the AE began on or after the date of the first dose of the active treatment extension period study medication and ≤30 days after the last dose of active treatment extension study treatment. Treatment emergent alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) ≥3 x upper limit of normal (ULN) and potential Hy's law cases were considered AEs of special interest and were followed until resolution and adjudicated by an external Hepatic Event Adjudication Committee. Safety evaluations were collected during the double-blind treatment period, active treatment extension period, and the safety follow-up period. An independent data safety monitoring board reviewed safety data regularly and provided oversight throughout the duration of the study.
Statistical analyses
A sample size of 130 participants per treatment group was estimated to provide approximately 88%, 94% or 99% power to show statistically significant improvement between each of the three atogepant doses (10 mg, 30 mg, or 60 mg) and placebo for the primary efficacy endpoint, respectively. Assumptions in this sample size calculation were based on the ADVANCE study in a US adult population (19). The dropout rate was assumed to be approximately 5.4% for the double-blind treatment period based on dropout rates observed in Japanese trials conducted in patients with EM (26–29).
Safety Population 1 was defined as all participants who received ≥1 dose of study drug during the double-blind treatment period. Safety Population 2 was defined as all participants who received ≥1 dose of study drug during the active treatment extension period. The modified intent-to-treat (mITT) population was defined as all randomized participants who received ≥1 dose of study drug and had ≥1 evaluable postbaseline four-week period of eDiary data. In order to be randomized, a participant had to be in the baseline period for ≥28 days and report eDiary data for ≥20 days during the baseline period. If <28 days of baseline data were reported, the number of headache days and other counting variables for baseline were prorated to standardize the count to a 28-day equivalent. If any postbaseline eDiary period for a participant had ≥14 but <28 days of reported data during a four-week period (Weeks 1–4, 5–8, or 9–12), the prorated approach for 28-day equivalent figures was used. Any four-week period with <14 days of completed eDiary data were considered missing. This prorating approach was applied to all efficacy analyses of eDiary data unless otherwise stated.
The change from baseline in MMDs at each monthly period was analyzed using a mixed model for repeated measures (MMRM) with treatment group, visit, prior exposure (yes/no) to a migraine preventive treatment, and treatment group-by-visit interaction as categorical fixed effects. Baseline score and baseline-by-visit interaction as covariates were also included. An unstructured covariance matrix was used to model the covariance of within-participant repeated measurements. The Kenward-Roger approximation was used to estimate the denominator degrees of freedom. The analysis was performed based on all postbaseline values using only the observed cases without imputation of missing values. Pairwise contrasts in the MMRM model were used to make the pairwise comparisons of each atogepant dose to placebo.
The secondary efficacy endpoints for headache days, acute medication use days, MSQ v2.1, and AIM-D were analyzed in a similar manner as the primary endpoint. The 50% responder rate endpoint was analyzed by a logistic regression model. This model assumed a binary distribution for the response and used a logit link. The analysis model included treatment group, prior exposure (yes/no) to migraine preventive treatment with proven efficacy, and baseline monthly migraine days.
All safety analyses were performed using Safety Populations 1 and 2. The safety parameters included AEs, clinical laboratory evaluations, vital sign measurements, ECG parameters, and the C-SSRS. For each of the clinical laboratory, vital sign, and ECG parameters, the last non-missing safety assessment before the first dose of treatment was used as the baseline for all analyses of that safety parameter. Continuous variables were summarized by the number of participants, mean, and standard deviation (SD). Categorical variables were summarized by number and percentage of participants.
Statistical analyses for the primary endpoint were calculated by adjusting for multiple comparisons using the Hochberg procedure with the family-wise type I error rate being controlled at a 0.05 level (2-sided). No multiplicity adjustments were provided for the secondary endpoints. Statistical analyses were performed using SAS software, version 9.4 or newer (SAS Institute, Cary, NC, USA).
Results
Participants
This study screened 807 participants for eligibility and enrolled a total of 524 participants. Safety Population 1 included 523 participants (placebo, N = 134; atogepant 10 mg, N = 126; 30 mg, N = 131; 60 mg, N = 132). The mITT population included 521 participants (placebo, N = 133; atogepant 10 mg, N = 127; 30 mg, N = 130; 60 mg, N = 131). Safety Population 2 included 511 participants (atogepant 10 mg, N = 169; 30 mg, N = 170; 60 mg, N = 172). Baseline demographics in Safety Population 1 were similar between treatment groups. Most participants were female (437/523, 83.6%), with a mean age of 43.0, a mean body mass index of 22.5 kg/m2, and 100% were Japanese (Table 1). Baseline demographics for Safety Population 2 were similar to Safety Population 1 (Online Supplemental Table 2). Most treated participants completed the 12-week double-blind treatment (511/523, 97.7%) and 12-week active treatment extension treatment (502/524, 95.8%). The most common reasons for study treatment discontinuation during the 12-week double-blind treatment period were adverse events (two participants receiving atogepant 30 mg, one receiving placebo, one receiving atogepant 10 mg, and one receiving atogepant 60 mg), withdrawal by participant (two participants receiving atogepant 30 mg, one receiving placebo, and one receiving atogepant 10 mg), lost to follow-up (one participant receiving placebo), protocol deviation (one participant receiving atogepant 30 mg), and other (one participant receiving atogepant 10 mg). The most common reasons for study treatment discontinuation during the 12-week active treatment extension period were adverse events (two participants receiving atogepant 60 mg, one receiving atogepant 10 mg), withdrawal by participant (two participants receiving atogepant 10 mg, one receiving atogepant 60 mg), other (one participant receiving atogepant 30 mg, one receiving atogepant 60 mg), and lost to follow-up (one participant receiving atogepant 60 mg) (Figure 1).

Study flow diagram. A participant is considered to have completed the double-blind study treatment if they have a Visit 5 date and received treatment for the active treatment extension period. A participant is considered to have completed the active treatment extension period if they have a Visit 8 date. A participant is considered to have completed the study if they have completed all regular study visits including the safety follow-up visit. *During the 12-week double-blind treatment period, one participant in the atogepant 10 mg QD group received study treatment other than the randomized study treatment. †One participant in the atogepant 60 mg group never received any study treatment. ‡During the 12-week active treatment extension period, one participant in the atogepant 60 mg QD group received study treatment other than the randomized study treatment. mITT, modified intent-to-treat; QD, once daily.
Baseline demographics and clinical characteristics (Safety Population 1).
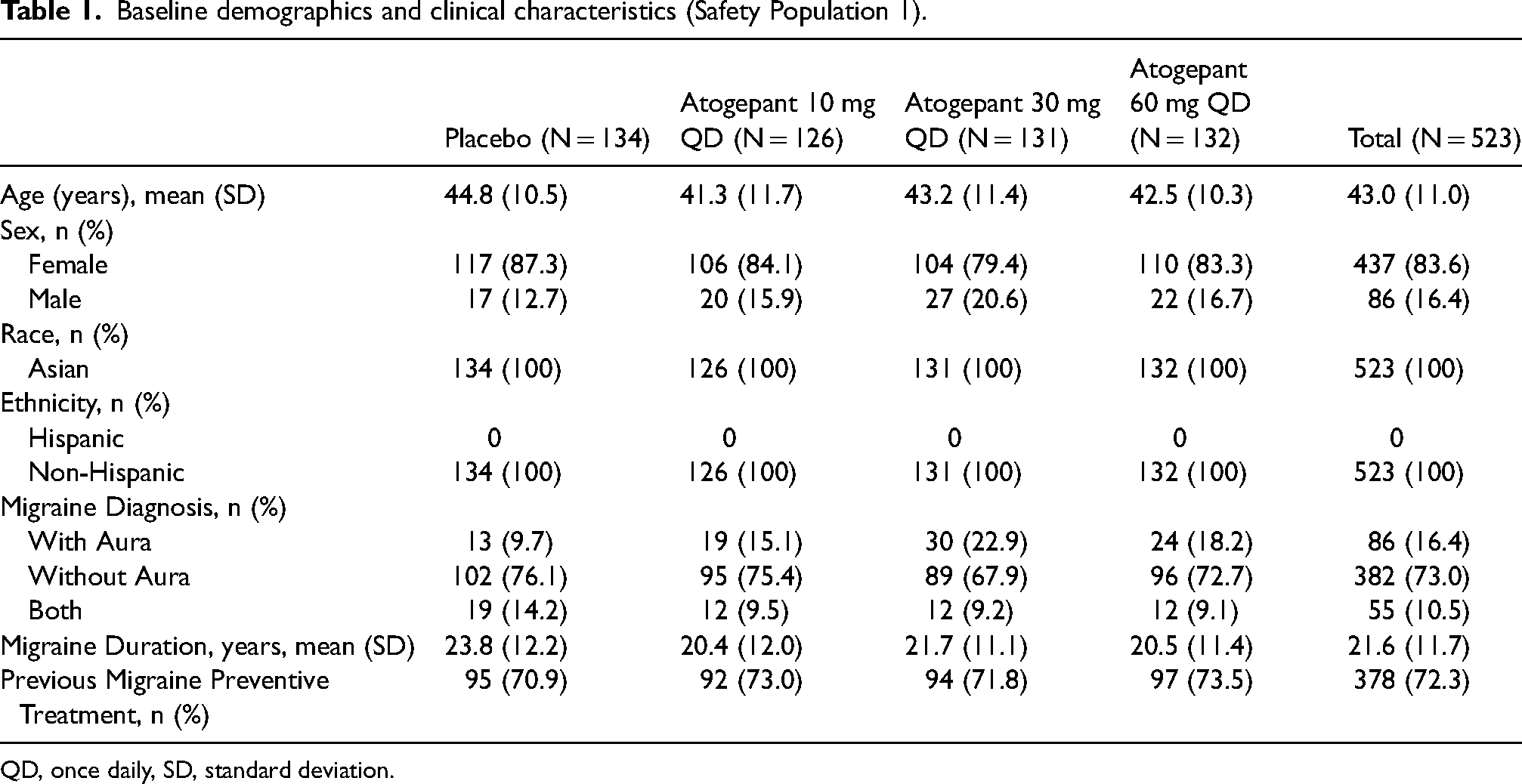
QD, once daily, SD, standard deviation.
Efficacy outcomes
Atogepant-treated participants had greater reductions from baseline in mean MMDs compared with placebo across the 12-week double-blind treatment period. The least square mean difference (LSMD) from placebo was −1.57 (95% CI, −2.24, −0.89) for atogepant 10 mg, −1.90 (95% CI, −2.57, −1.22) for 30 mg, and −2.10 (95% CI, −2.78, −1.43) for 60 mg. All comparisons demonstrated statistically significant reductions in mean MMDs compared with placebo (p < 0.0001) (Table 2). All doses of atogepant demonstrated a reduction in MMDs as early as Weeks 1–4 and persisted through Weeks 9–12 (Figure 2). Across the 12-week, double-blind treatment period, a greater proportion of participants in the atogepant groups achieved a ≥50% reduction in mean MMDs compared with placebo. A reduction of ≥50% was achieved by 15.0% of participants receiving placebo, 40.9% receiving atogepant 10 mg, 42.3% receiving 30 mg, and 50.4% receiving 60 mg. The odds ratio (95% CI) of a ≥50% reduction compared with placebo was 4.0 (2.19, 7.20) for atogepant 10 mg, 4.2 (2.33, 7.60) for 30 mg, and 5.9 (3.27, 10.61) for 60 mg (all comparisons nominal p < 0.0001) (Table 2).

Least square mean change from baseline in monthly migraine days at weeks 1–4, 5–8, and 9–12 in the 12-week double-blind treatment period (mITT population). CI, confidence interval; LS, least square, LSMD, LS mean difference; mITT, modified intent-to-treat; SE, standard error.
Primary and secondary endpoints across the 12-week double-blind treatment period (mITT population).

AIM-D, Activity Impairment in Migraine-Diary; CI, confidence interval; LS, least squares; LSMD, LS mean difference; mITT, modified intent-to-treat; MSQ v2.1, Migraine-Specific Quality of Life Questionnaire version 2.1; PDA, Performance of Daily Activities; PI, Physical Impairment; QD, once daily; RFR, Role Function-Restrictive; SD, standard deviation; SE, standard error.
Atogepant-treated participants had greater reductions from baseline in mean MHDs compared with placebo across the 12-week double-blind treatment period. The LSMD from placebo was −2.16 (95% CI, −2.90, −1.42) for atogepant 10 mg, −2.30 (95% CI, −3.04, −1.57) for 30 mg, and −2.49 (95% CI, −3.22, −1.75) for 60 mg (all comparisons nominal p < 0.0001). Additionally, atogepant-treated participants had greater reductions from baseline in mean monthly acute medication use days compared with placebo across the 12-week double-blind treatment period. The LSMD from placebo was −2.12 (95% CI, −2.80, −1.44) for atogepant 10 mg, −2.33 (95% CI, −3.01, −1.66) for 30 mg, and −2.48 (95% CI, −3.15, −1.81) for 60 mg (all comparisons nominal p < 0.0001) (Table 2).
The LSMD from placebo at Week 12 in the MSQ v2.1 RFR domain score was 7.06 (95% CI, 3.65, 10.47) for atogepant 10 mg, 8.86 (95% CI, 5.47, 12.25) for 30 mg, and 9.23 (95% CI, 5.86, 12.60) for 60 mg (all comparisons nominal p < 0.0001), which exceeded the threshold for clinical meaningfulness (between-group MID of ≥3.2 points), demonstrating improvement from baseline in daily social and work-related functioning compared with placebo (Table 2). Results for the MSQ v2.1 RFR domain at Weeks 4 and 8 were consistent with all doses of atogepant (Online Supplemental Figure 2). Exploratory analyses of the MSQ v2.1 RFP and EF domain scores demonstrated improvement with atogepant 30 mg and 60 mg compared with placebo as early as Week 4 and persisted through Week 12 (Online Supplemental Figure 2). At Week 12, the LSMD between all doses of atogepant compared with placebo in the MSQ v2.1 RFP domain, and atogepant 60 mg compared with placebo in the EF domain, reached the between-group MIDs and thus are clinically meaningful improvements.
The LSMD from placebo across 12 weeks in AIM-D PDA domain score was −2.39 (95% CI, −3.37, −1.40) for atogepant 10 mg, −2.96 (95% CI, −3.94, −1.98) for 30 mg, and −2.55 (95% CI, −3.53, −1.57) for 60 mg (all comparisons nominal p < 0.0001), demonstrating improvement from baseline in the ability to perform daily functions compared with placebo. The LSMD from placebo across 12 weeks in AIM-D PI domain score was −2.20 (95% CI, −3.17, −1.23) for atogepant 10 mg, −2.77 (95% CI, −3.73, −1.81) for 30 mg, and −2.37 (95% CI, −3.33, −1.41) for 60 mg (all comparisons nominal p < 0.0001), demonstrating improvement from baseline in physical impairment compared with placebo (Table 2). AIM-D total score, PDA domain score, and PI domain score demonstrated improvement with all atogepant doses compared with placebo as early as Weeks 1–4 and persisted through Weeks 9–12 (Online Supplemental Figure 3).
Treatment-emergent adverse events
During the 12-week double-blind treatment period, TEAEs occurred in 46.3% (62/134), 45.2% (57/126), 38.9% (51/131), and 43.2% (57/132) of participants in the placebo, atogepant 10 mg, 30 mg, and 60 mg treatment groups, respectively (Table 3). TEAEs that occurred in ≥5% of participants in Safety Population 1 were constipation (placebo, 4.5%; atogepant 10 mg, 4.0%; 30 mg, 7.6%; 60 mg, 9.8%) and nasopharyngitis (placebo 10.4%, atogepant 10 mg, 5.6%, 30 mg, 9.9%, 60 mg, 6.1%) (Table 4). During the 12-week active treatment extension period, TEAEs occurred in 45.6% (77/169), 46.5% (79/170), and 43.6% (75/172) of participants in the atogepant 10 mg, 30 mg, and 60 mg treatment groups, respectively (Table 3). One TEAE (nasopharyngitis) occurred in ≥5% of participants in Safety Population 2 (atogepant 10 mg, 10.7%, 30 mg, 11.8%, 60 mg, 11.0%) (Online Supplemental Table 3). TEAEs were consistent across the full study duration (Weeks 1–24) (Table 4). No deaths were reported during the study. There was one serious TEAE (prinzmetal angina) in the atogepant 10 mg treatment group in Safety Population 1 during the double-blind treatment period, and it was considered not related to atogepant treatment by the investigator. There were two serious TEAEs (oropharyngeal cancer, hemorrhoids) in the atogepant 10 mg treatment group and one serious TEAE (anemia) in the atogepant 30 mg treatment group in Safety Population 2 during the active treatment extension period, and these were not considered related to atogepant treatment by the investigator. Very few participants discontinued treatment due to TEAEs. During the double-blind treatment period, there were two participants with TEAEs leading to treatment discontinuation in the atogepant 30 mg treatment group (gastroenteritis, alopecia), and one participant each in the placebo (vaginal hemorrhage), atogepant 10 mg (prinzmetal angina), and 60 mg treatment groups (decreased appetite and vomiting) in Safety Population 1. The TEAEs, gastroenteritis, vaginal hemorrhage, decreased appetite, and vomiting, were assessed as reasonable possibility to be related to study treatment. The TEAEs, alopecia and prinzmetal angina, were assessed as having no reasonable possibility of being related to study treatment. During the active treatment extension period, there were two participants with TEAEs leading to treatment discontinuation in the atogepant 60 mg treatment group (nausea, neck pain) and one in the atogepant 10 mg treatment group (oropharyngeal cancer) in Safety Population 2 (Table 3). The TEAEs, nausea and neck pain, were assessed as having a reasonable possibility of being related to study treatment. The TEAE, oropharyngeal cancer, was assessed as having no reasonable possibility of being related to study treatment.
Treatment-emergent adverse events during the 12-week double-blind treatment period (Safety Population 1) and 12-week active treatment extension period (Safety Population 2).

QD, once daily.
Treatment-emergent adverse events (≥2%) in the 12-week double-blind treatment period (weeks 1–12) and double-blind treatment period and active treatment extension period (weeks 1–24) (Safety Population 1).

QD, once daily.
TEAEs in bold represent those occurring ≥5%.
Clinical laboratory evaluation and vital signs
There were no clinically meaningful differences across treatment groups in the proportion of participants with postbaseline potentially clinically significant hematology, chemistry, or urinalysis values in Safety Populations 1 and 2. Overall, hepatic function laboratory values of clinical interest were comparable across treatment groups for mean changes from baseline to the end of the 12-week double-blind treatment period. During the 12-week double-blind treatment period, no participants had an elevation of ALT or AST ≥3 × ULN. During the 12-week active treatment extension period through the end of the study, three participants had an ALT or AST of ≥3 × ULN; two during the active treatment extension period (one in the atogepant 10 mg QD treatment group who had received placebo in the double-blind treatment period and one in the atogepant 60 mg treatment group who had received placebo in the double-blind treatment period), and one at the Week 28/Follow-up visit in the atogepant 10 mg QD treatment group. ALT or AST elevations had returned to <1 × ULN within four weeks. These three cases were evaluated by an external Hepatic Event Adjudication Committee, and all were assessed as possibly related to study treatment. No participant had laboratory values that met criteria for Hy's Law (Online Supplementary Table 4).
Discussion
There remains a high unmet need in the Japanese population for newer migraine preventive treatments, and we report here the first results of atogepant for the preventive treatment of EM in Japanese participants. The RELEASE trial met the primary endpoint and all doses of atogepant demonstrated a statistically significant reduction in mean MMDs across the 12-week double-blind treatment period compared with placebo. This reduction in MMDs was observed as early as Weeks 1–4 and was consistent through Weeks 9–12. Additionally, all doses of atogepant demonstrated reductions in mean MHDs, reductions in mean monthly acute medication use days, and higher proportions of participants achieving a ≥50% reduction in mean MMDs across the 12-week double-blind treatment period. Atogepant treatment also demonstrated improvements in key functional outcomes compared with placebo. Atogepant was safe and well tolerated across the two 12-week treatment periods with very few participants discontinuing treatment due to TEAEs (one participant each in the placebo, atogepant 10 mg, and 60 mg groups, and two participants in the atogepant 30 mg group). No new safety signals were observed in the Japanese population. The safety profile of atogepant in this trial was consistent with the established safety profile in global trials (18–20).
The unmet need for newer migraine preventive treatments in Japan with demonstrated efficacy and safety has led to phase 2 and 3 trials investigating the use of other CGRP targeted migraine preventive treatments, including injectable CGRP mAbs for the preventive treatment of EM. Erenumab, galcanezumab, and fremanezumab all demonstrated a reduction in MMDs compared with placebo in randomized, double-blind, placebo-controlled trials in a Japanese population (26–29). Here we report that the oral CGRP receptor antagonist, atogepant, demonstrated statistical superiority to placebo in the reduction of mean MMDs across the 12-week, double-blind treatment period. A discrete choice experiment of 400 participants in Japan found that the method of delivery was the most important treatment attribute for patients selecting a migraine treatment. Additionally, when selecting a preventive migraine treatment, oral preventive treatments were preferred over injectable preventive treatments (30). This study suggests that oral atogepant may be a preferred preventive treatment option compared to injectable treatments in Japan.
The placebo-corrected treatment differences for the primary and secondary endpoints in this Japanese trial are consistent with those in the US based and global atogepant trials (18–20). The LSMD between atogepant 60 mg QD and placebo in mean MMDs across the 12-week treatment period in the atogepant phase 2b/3 study in EM (CGP-MD-01) (20), phase 3 study in EM (ADVANCE) (19), and phase 3 study in EM with prior preventive treatment failure (ELEVATE) (18) were −0.7, −1.7, and −2.4 days, respectively. The LSMD from placebo in MMDs reported here for RELEASE was −2.1 days. The atogepant 60 mg QD dose demonstrated the greatest improvement in efficacy and functional outcomes with no clinically relevant increase in TEAEs. The 60 mg QD dose is an approved dose for the preventive treatment of migraine in the US and EU (16,17). Currently, patients in Japan frequently switch or discontinue oral preventive treatments due to poor tolerability and suboptimal efficacy (12). Taken together, the consistency of the outcomes observed here with those of the US based and global atogepant trials support the use of oral atogepant as a preventive treatment option in Japan.
The RELEASE trial comprised mostly female participants. Additionally, participants were excluded if they used opioids or barbiturates ≥2 days per month in the three months before Visit 1 or during the baseline period, and those who did not have a response to >4 preventive treatments. Participants with comorbid conditions outlined in the methods and those who are pregnant, planning on becoming pregnant, or are currently lactating were excluded. Collectively, this may limit the generalizability of the results to the full patient population. The 24-week treatment period is short, and analyses of longer treatment durations in Japanese populations are needed. A phase 3, 52-week, open-label extension trial evaluating the safety, tolerability, and efficacy of atogepant (NCT04437433) will evaluate long-term outcomes of atogepant treatment in Japanese participants with EM and chronic migraine.
Conclusions
This is the first report of atogepant for the preventive treatment of EM in Japanese participants. Across the 12-week double-blind treatment period, atogepant treatment demonstrated a statistically significant reduction in mean MMDs compared with placebo. Additionally, atogepant treatment demonstrated a reduction in mean MHDs and monthly acute medication use days, an increase in the proportion of participants achieving a ≥50% reduction in mean MMDs, and improvements in functional outcomes compared with placebo. Atogepant was safe and well tolerated across the two 12-week treatment periods in Japanese participants. No new safety signals were observed in the Japanese population, and the safety profile was consistent with previous global trials in EM (18–20).
Article highlights
Atogepant treatment demonstrated statistically significant reductions in mean MMDs compared with placebo across the 12-week double-blind treatment period in Japanese participants with episodic migraine
Atogepant treatment demonstrated improvements in all secondary efficacy and functional outcomes compared with placebo
Safety across the two 12-week treatment periods was consistent with the known safety profile of atogepant; no new safety signals were identified
Supplemental Material
sj-docx-1-cep-10.1177_03331024251374569 - Supplemental material for Atogepant for the preventive treatment of episodic migraine in Japanese participants: A phase 2/3, randomized, double-blind, placebo-controlled trial with an active treatment extension (RELEASE)
Supplemental material, sj-docx-1-cep-10.1177_03331024251374569 for Atogepant for the preventive treatment of episodic migraine in Japanese participants: A phase 2/3, randomized, double-blind, placebo-controlled trial with an active treatment extension (RELEASE) by Yasuhiko Matsumori, Hiroshi Yamada, Yoshishige Nagaseki, Kazutaka Shimizu, Krisztian Nagy, Ryotaro Matsuzawa, Tetsuya Otani, Molly Yizeng He, Hua Guo, Gina Ahmadyar, and Takao Takeshima in Cephalalgia
Supplemental Material
sj-docx-2-cep-10.1177_03331024251374569 - Supplemental material for Atogepant for the preventive treatment of episodic migraine in Japanese participants: A phase 2/3, randomized, double-blind, placebo-controlled trial with an active treatment extension (RELEASE)
Supplemental material, sj-docx-2-cep-10.1177_03331024251374569 for Atogepant for the preventive treatment of episodic migraine in Japanese participants: A phase 2/3, randomized, double-blind, placebo-controlled trial with an active treatment extension (RELEASE) by Yasuhiko Matsumori, Hiroshi Yamada, Yoshishige Nagaseki, Kazutaka Shimizu, Krisztian Nagy, Ryotaro Matsuzawa, Tetsuya Otani, Molly Yizeng He, Hua Guo, Gina Ahmadyar, and Takao Takeshima in Cephalalgia
Footnotes
Acknowledgements
AbbVie and the authors thank all the trial investigators and the patients who participated in this clinical trial. Medical writing assistance was provided to the authors by Brian Neel, PhD, of AbbVie (North Chicago, IL, USA), and editorial assistance was provided to the authors by Angela T. Hadsell, BA, of AbbVie (North Chicago, IL, USA), and was funded by AbbVie.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: YM reports personal consultancy fees from Amgen, Astellas, BioPharma K.K., Daiichi Sankyo Co., Ltd, Eli Lilly Japan K.K., and Otsuka Pharmaceutical Co., Ltd. HY has received speaker honoraria from Daiichi Sankyo Co., Ltd, Eli Lilly Japan K.K., and Otsuka Pharmaceutical Co., Ltd. YN reports personal consultancy fees from Amgen, K.K., Daiichi Sankyo Co., Ltd, Eli Lilly Japan K.K., and Otsuka Pharmaceutical Co., Ltd. KS has received speaker honoraria from Amgen Inc. and Daiichi Sankyo Co., Ltd. TT has been on the speakers’ bureau for Amgen K.K., Daiichi Sankyo Co., Ltd, Eli Lilly Japan K.K., and Otsuka Pharmaceutical Co., Ltd, and has received research funding/collaborative research expenses from AbbVie GK., Amgen K.K., Eli Lilly Japan K.K., Eisai Co., Ltd, Lundbeck Japan K.K., and Pfizer Japan Inc. TT also acted as an advisor to Hedgehog MedTech, Inc., Sawai Pharmaceutical Co., Ltd, and TEIJIN Pharma Ltd. RM, TO, MYH, HG, GA, and KN are employees of AbbVie and may own AbbVie stock.
Data availability statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan, and execution of a Data Sharing Agreement. Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: ![]() then select “Home”.
then select “Home”.
Standard protocol approvals,registration,and patient consents
The trial design was in compliance with the International Headache Society guidelines for controlled trials of preventive treatment of migraine attacks in EM (![]() ). The trial was approved by a local or central Institutional Review Board at each participating institution and conducted in accordance with the International Conference for Harmonisation guidelines, applicable regulations, and the Declaration of Helsinki. Participants provided written informed consent before screening. The study is registered with ClinicalTrials.gov (NCT05861427).
). The trial was approved by a local or central Institutional Review Board at each participating institution and conducted in accordance with the International Conference for Harmonisation guidelines, applicable regulations, and the Declaration of Helsinki. Participants provided written informed consent before screening. The study is registered with ClinicalTrials.gov (NCT05861427).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.