
Abstract
Objective
We investigated whether greater occipital nerve injection (GON injection) with 80 mg of methylprednisolone at the onset of a cluster headache episode would reduce attack frequency faster than standard therapy with verapamil alone, and reduce the need for verapamil and the risk of adverse events (AEs).
Methods
This was an investigator-initiated, randomised, double-blind, 12-week clinical trial. Participants received GON injection with 80 mg of methylprednisolone (n = 36) or placebo (n = 34) within two weeks (median) after the onset of a cluster episode, followed by standard verapamil therapy and e-diary monitoring. The primary endpoint was the mean daily dose of verapamil over the entire 12-week study period. Key secondary endpoints were reduction in the mean daily dose of verapamil over the first four weeks and attack frequency reduction in the first week.
Results
In the verum vs. placebo group, the mean daily dose of verapamil during the total 12-week study period did not differ (232 ± 188 mg vs. 244 ± 143 mg; Δ = 12 mg, 95% confidence interval (CI) = −68 to 92; p = 0.230). However, exploratory analysis of the secondary endpoints showed a lower verapamil dose in the first four weeks in the methylprednisolone group compared to placebo (227 ± 126 mg vs. 287 ± 107 mg; mean Δ 60 mg; 95% CI = −4 to −116), as was the median number of attacks at week 1 (7 (interquartile range = 2–11.75) vs. 10 (interquartile range = 6–17.5); 95% CI = −1.0 to −8.0), the mean attack intensity at week 1 (5.7 ± 1.9 vs. 6.6 ± 1.8; 95% CI = 0.0–1.8) and throughout the 12-week study period (5.0 ± 1.8 vs. 5.9 ± 1.9; 95% CI = 0.01–1.8), and the number of days with adverse events (455/2520 (18%) vs. 605/2850 (21%); p < 0.01). There were no serious AEs.
Comclusions
This study failed to establish its primary endpoint. However, exploratory analysis of the secondary endpoints revealed that GON injection with 80 mg of methylprednisolone at the beginning of a cluster headache episode followed by standard therapy verapamil is a safe transitional treatment that provides faster reduction in attack frequency and intensity than verapamil alone, decreases the mean verapamil dose over the first four weeks with consequently fewer adverse events in the first four weeks after the injection.
Trial Registration
This study is registered on Clinicaltrials.gov with registration number NCT04014634 at 08-07–2019. First inclusion was on 30-07-2019.
This is a visual representation of the abstract.
Keywords
Introduction
Episodic cluster headache (ECH) is characterised by recurrent episodes lasting several weeks to months with attacks of excruciating strictly unilateral headache for 15–180 minutes and ipsilateral autonomic symptoms in the face and/or restlessness. Typically, episodes start with a few attacks that then become increasingly frequent up to 8–10 per day to slowly disappear again after a plateau phase. These periods with attacks alternate with longer periods of several months to many years without attacks (1,2–5), and (ii) prevention of attacks, usually with off-label verapamil, a calcium channel blocker primarily designed to control cardiovascular symptoms (5–6). To reduce the risk of adverse events (AEs), dosing should be titrated slowly over several weeks with frequent electrocardiogram (ECG) monitoring. It may therefore take several weeks before effective doses are achieved, during which patients continue to suffer from frequent incapacitating headaches (5,8). A faster attack-preventive effect at lower doses of verapamil with fewer AEs would significantly improve the treatment of ECH. High doses of oral prednisone can also effectively prevent cluster headache (CH) attacks, but because of the very high risk of a wide range of SAEs, they are rarely prescribed and then only for a short period of time with consequently only short efficacy.
Greater occipital nerve injection (GON injection) with steroids can reduce attacks in CH (9–18) However, as highlighted in a recent systematic review and meta-analysis (19,20), much of the research is of poor methodological quality and subgroup analysis for ECH could not be performed due to the low number of participants. Moreover, ECH patients often respond differently to treatment than chronic CH (CCH; no attack-free periods) patients (7,19,20). Important limitations include that the studies are often small and open-label (9–11,13,14,16), participants had different prophylactic co-medications in different doses and, importantly, administration of GON injection at non-standardised times, often many weeks after the onset of a cluster episode (10,15,17–21). Although “peri occipital nerve infiltration” probably is a more accurate description of and more appropriate term for the procedure, “GON block or injection” is the most commonly used term. To avoid confusion we shall use this more conventional term in this article.
In this randomised controlled trial, we aimed to investigate whether GON injection with 80 mg of methylprednisolone at the beginning of a cluster episode prior to standard therapy with escalating doses of daily oral verapamil, compared with usual therapy with oral verapamil alone, results in a more rapid decrease in attack frequency and a lower required dose of verapamil and therefore fewer AEs.
Methods
Study design
The CHIANTI trial (Cluster Headache: peri-occipital nerve Infiltration As New Treatment Intervention) is an investigator-initiated, multicentre, randomised, double-blind, placebo-controlled clinical trial in which participants either received GON injection with 80 mg of methylprednisolone or placebo, followed by standard titration of verapamil in the subsequent weeks. Participants were followed for 12 weeks during which they completed a daily e-diary. In addition, there were consultations by phone once or twice per week.
Written informed consent was obtained from all participants according to the Declaration of Helsinki and the study protocol was approved by the ethical committee of the LUMC (METC-LDD; Protocol number P18.242) and each participating centre's local ethics committee. This study is registered on Clinicaltrials.gov with registration number NCT04014634 at 08-07-2019. First inclusion was on 30 July 2019 and follow-up lasted until 19 November 2022.
Participants
Known patients with ECH were recruited from one academic and five non-academic headache clinics in the Netherlands and were asked to contact the participating centre at the onset of a cluster episode. After pre-screening by telephone, they were invited for the first study visit at which eligibility was confirmed by a study neurologist. Patients were then formally included and received GON injection with 80 mg of methylprednisolone or placebo. Newly diagnosed patients could also be included, as long as they were at the beginning of a cluster episode.
Participants were aged at least 18 years with ECH (1) with three or more attacks in the previous three days and less than four weeks from the onset of a cluster episode and currently free from prophylactic treatment. Exclusion criteria were contraindication for steroids or verapamil, prophylaxis for other headache types, use of anticoagulants, known bleeding disorder, and historically cluster episodes for less than four weeks.
Randomisation and masking
Participants were randomised (1:1) via Castor Electronic Data Capture. Block randomisation with blocks of two, four and six was used and stratified by participating centres. The hospital pharmacist or an independent neurologist (CWZ Hospital, Nijmegen, the Netherlands) prepared the syringe on site according to standard protocol. The syringe was then covered with aluminum foil to maintain blinding. This was lifted for RB and EZ only after all participants completed the trial and the database was cleaned.
Treatment
GON injection
GON injection with 2 ml of suspension containing 80 mg of methylprednisolone or saline was administered ipsilateral to the side of the pain during attacks in a blinded fashion by RB, WM, OG or RF. The injection site was based on visual and palpable landmarks (one-third of the line between the occipital protuberance and the mastoid process) (15).
Verapamil
Because of the extremely painful attacks and high disease burden, it was deemed unethical to treat participants with placebo GON injection alone, without any other form of prophylactic treatment. Although effective attack therapy is available, the acute effect is certainly not immediate. Placebo-treated participants would still have to suffer terrible pain for at least 15 minutes, if not longer, with each attack they have before being completely pain-free. Moreover, sumatriptan treatment is formally limited to a maximum of two injections per day. Most patients have significantly more attacks per day that then have to be treated with significantly less effective treatments. Hence, in addition to GON injection, all participants received standard prophylaxis with verapamil 120 mg extended-release in escalating doses, which could be continued after the study.
Attack treatment
Attacks could be treated with 3 or 6 mg of subcutaneous sumatriptan, 20 mg of sumatriptan nasal spray, inhalation of pure oxygen, or a combination of these.
Verapamil dose escalation
Participants were contacted by telephone every three days, except weekends. Participants who were attack-free for one week were contacted by the study team for two additional three-day intervals. If the participants were then still attack-free, weekly contacts were scheduled. The dose was increased with 120 mg if participants were not attack free on the day of the consultation and the day before, and adverse events were absent or tolerable. The dose was maintained if participants had not had attacks on the day of the consultation or the day before, or if the participant experienced side effects or did not want to increase the dose. The dose of verapamil was reduced by 120 mg per week if the participant had not had any attack for at least four weeks, the participant subjectively felt that the cluster period was over, or if side effects became too bothersome (see supplementary material, Figure S1). Preventive treatment other than verapamil was not allowed during the 12-week study period. If attacks persisted for one week and the verapamil dosage could not be increased because the participants was already using the maximum dose of verapamil (720 mg) or because side effects were too severe, and the participant requested additional preventive treatment, participants were offered topiramate, lithium or oral prednisone as rescue medication. A participant was then considered to be a drop-out and their data was analysed using the last observation carried forward principle and imputed using multiple imputations.
Procedures
According to standard clinical procedures in the Netherlands, ECGs were performed at the start of treatment and at daily doses of 360 mg and 720 mg. Participants completed a daily e-diary that included all study parameters: occurrence and intensity of attacks (all participants were instructed to report attacks they considered to be cluster attacks), use of acute attack medication, verapamil dosage, other types of headache and general well-being on a seven-point Likert scale. All data remained confidential and were masked from the entire research team during the study.
Outcomes
The primary outcome was the mean daily dose of verapamil over the entire 12-week study period. The key secondary endpoint were the mean daily dose of verapamil over the first four weeks and the median number of attacks per day during the first week. Other prespecified secondary endpoints were: the peak dose verapamil, premature termination of the study due to needs for prophylactic escape medication and the median number of days to remission, defined as seven consecutive days without attack.
Prespecified tertiary outcomes are listed in the supplementary material Doc. S1.
Sample size calculation
A previous trial showed a mean dose of verapamil in the placebo group of 546 mg (standard deviation 180 mg) (17) To detect a 30% decrease in total verapamil dose during days 1–28 with a power of 90% and a 5% bilateral significance threshold, a sample size of 52 participants (26 per treatment group) was needed. We included 35 participants per treatment group to allow for dropouts.
Statistical analysis
All analyses were performed in the intention-to-treat (ITT) population, which consisted of all participants who were randomised and had received a GON-injection. Data are presented as the mean ± SD or as the median and interquartile range (IQR) when appropriate for continuous variables and as number and percentage for categorical variables. Adverse events are presented as a rate (person per day) and as the number of participants who reported the event with percentage per group.
The primary endpoint was analysed with Student’s t-test. Secondary endpoints were analysed with Student’s t-test, a Wilcoxon rank test, a log-rank test for the Kaplan–Meier curve and a rate ratio test for the AEs, when appropriate. We used the primary endpoint as a “gatekeeper” endpoint, testing at a significance level of 0.05. If the primary endpoint was met, the key secondary endpoint would start with a significance level of 0.05 and a Bonferroni correction for multiple testing would be used for the other secondary endpoints. If the primary endpoint was not met, all other endpoints would be considered exploratory. No correction for multiple testing was used in the tertiary analyses and the safety analyses, which should be considered exploratory.
Missing values for the primary outcome were imputed. Variations in the imputed datasets were analysed and pooled data from five different imputed datasets was used. Age, sex, attack frequency at baseline and daily attack frequency were used as predictors with predictive mean matching.
The study protocol and statistical analysis plan are available upon request.
Results
Of the 185 patients screened for eligibility, 70 participants were randomised, 36 in the methylprednisolone and 34 in the placebo group, and included in the primary ITT analysis (Figure 1). First inclusion was on 30 July 2019 and follow-up lasted until 19 November 2022. The trial ended after all required participants finished their follow-up. Reasons for exclusion are listed in Figure 1. No difference between treatment groups were observed in the baseline characteristics, except for a history of slightly longer cluster episodes in the methylprednisolone group (Table 1) which did not influence the primary results (r = 0.16; 95% confidence interval (CI) = −0.10 to 0.4).

Participant flowchart. CCH: chronic cluster headache.
Baseline characteristics.

GON: greater occipital nerve.
GON injection was administered at a median of two weeks after onset of the cluster episode (IQR = 1–3 weeks) in both groups (p = 0.232).
Primary endpoint
The mean daily dose of verapamil over the entire 12-week study period did not differ between groups. Methylprednisolone group: 232 ± 188 mg vs. 244 ± 143 mg; Δ = 12 mg (95% CI = −68 to 92; p = 0.230).
Key secondary endpoints
The mean daily dose of verapamil in the first four weeks was lower in the methylprednisolone group: 227 ± 126 mg vs. 287 ± 107 mg; Δ = 60 mg (95% CI = −4 to −116; p = 0.036) (Figure 2).

Mean daily verapamil dose over the first four weeks after greater occipital nerve (GON) injection. A lower mean daily dose of verapamil in weeks 1–4 was observed in the methylprednisolone group (227 ± 126 mg) than in the placebo group (287 ± 107 mg; difference = 60 mg, 95% confidence interval = −4 to −116; p = 0.036).
The median number of weekly attacks was lower during the first week in the methylprednisolone group (7 (IQR = 2–11.75) vs. 10 (IQR = 6–17.5); Δ = −3, 95% CI = −1.0 to −8.0; p = 0.016) (Figures 3 and 4; imputed data are provided in the supplemental material, Figure S2).

Median number of weekly attacks with interquartile range during the first four weeks after greater occipital nerve (GON) injection per group. *p< 0.05.
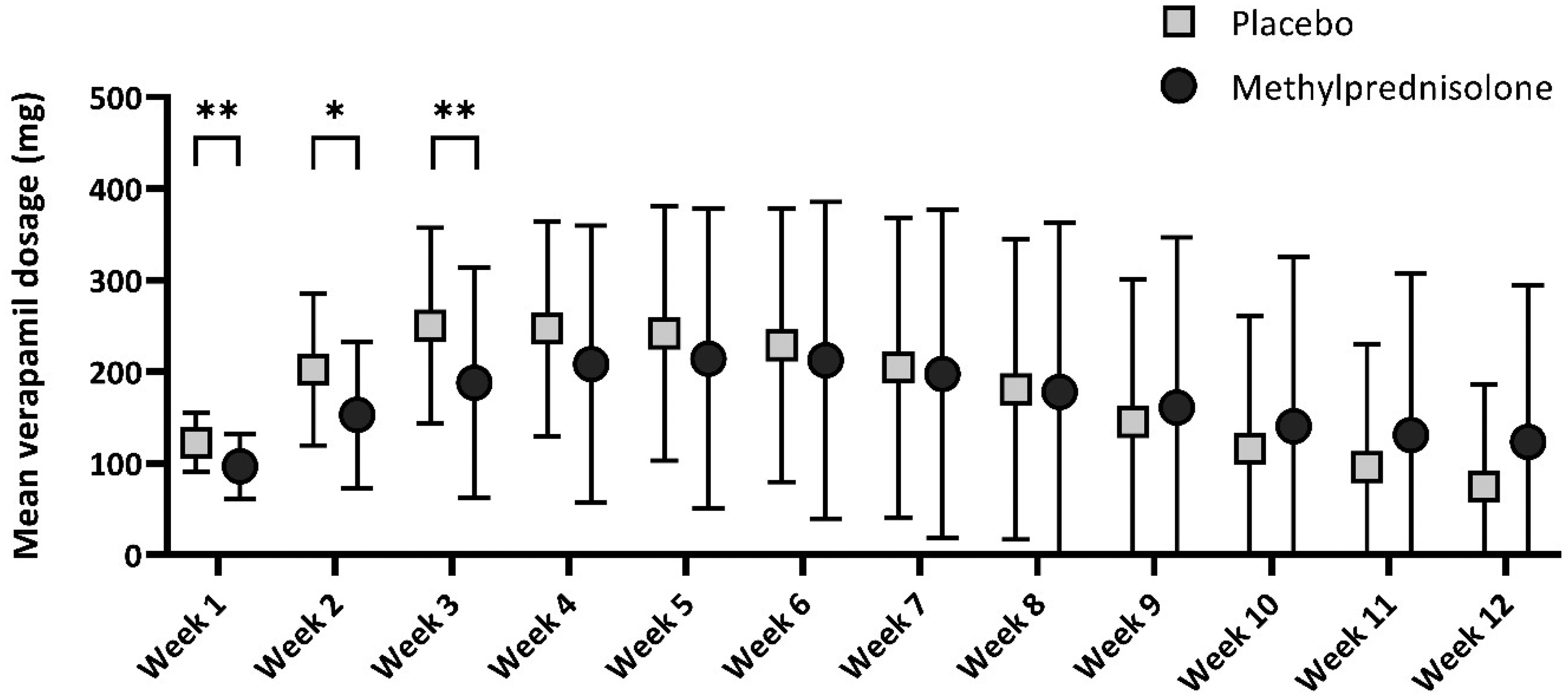
Mean verapamil dose with 95% confidence interval per day. *p ≤ 0.05, **p ≤ 0.01.
Other secondary endpoints
There were no differences for the mean number of days to remission (verum: 25.5 ± 16.6 vs. placebo: 27.3 ± 10.1; see supplemental material, Figure S3), premature termination of the trial (verum 12% vs. placebo 6%) and the peak dose of verapamil (verum: 360 ± 213 vs. placebo: 440 ± 146).
Tertiary endpoints
Tertiary endpoints are presented in the supplemental material (Table S1).
The mean attack intensity was lower in the methylprednisolone group in week 1 (5.7 ± 1.9 vs. 6.6 ± 1.8; 95% CI = 0.0–1.8) and over the entire 12-week study period (5.0 ± 1.8 vs. 5.9 ± 1.9; 95% CI = 0.01–1.8). Well-being was higher in the methylprednisolone group after seven days (6.4 ± 1.6 vs. 5.4 ± 2.0; 95% CI = −1.9 to −0.1).
The percentage of participants with attack freedom was higher in the methylprednisolone group after seven days (59% vs. 34%, p = 0.017), but not after 14 or 28 days (14 days: 69% vs. 61%, p = 0.51; 28 days: 81% vs. 77%, p = 0.73).
There were no differences for the mean daily dose of verapamil and the median number of weekly attacks over the entire 12-week study period, time to peak dose verapamil and sumatriptan or oxygen use.
Adverse events
Registration of AEs was completed for 5370/5880 (91%) follow-up days and presented in the supplemental material (Table S2). Days with AEs were less frequent in the methylprednisolone group (455/2520, 18% vs. 605/2850, 21%; p < 0.01), especially during the first four weeks (232/874, 27% vs. 340/992, 34%; p = 0.003). Most commonly reported AEs were tiredness and obstipation. In each group, one SAE occurred, which were deemed unrelated to study treatment.
Discussion
A single GON injection with 80 mg of methylprednisolone, administered within a median of two weeks after the onset of a cluster episode and followed by standard therapy with verapamil in escalating doses did not reduce the mean daily dose of verapamil over the entire 12-week study period. However, exploratory analysis of the secondary endpoints revealed that it did reduce the required dose of verapamil in the first four weeks and reduced the attack frequency in the first week. Furthermore, GON injection was well tolerated and safe, reduced attack frequency and intensity faster than verapamil alone, and reduced the verapamil-related adverse event rate over the first four weeks. Moreover, in the verum group, more participants were attack-free a week after GON injection and the overall well-being was higher despite the historical duration of cluster episodes in the methylprednisolone group being slightly longer. Although no effect was observed over de entire 12-week period, these results underline that GON injection with 80 mg of methylprednisolone at the beginning of a cluster period significantly improves the prophylactic treatment of CH with lower required doses of verapamil, consequently fewer adverse events in the first four weeks after the injection and reduced attack frequency and intensity faster than verapamil alone, confirming earlier retrospective data from our group (22).
We failed to achieve our primary endpoint, reduction in mean daily verapamil use over the entire 12-week period. The beneficial effect of GON injection was mainly visible in the first few weeks, after which the results for placebo and verum began to converge. After the entire 12-week study period, all differences have disappeared. This could mainly be attributed to the highly effective treatment with verapamil, and the earlier than anticipated convergence of the groups over time. Other explanations may be (i) a smaller and shorter effect of the GON injection than we had previously anticipated in combination with (ii) the natural transient disease course (which ends spontaneously after a certain period of time). However, we did achieve our key secondary endpoints: reduction in mean daily verapamil dose during the first four weeks, and attack reduction after one week, cementing the use of GON injection as a bridging therapy. With this study, we replicate the efficacy of GON injection in the first four weeks that was observed in a recent randomised controlled trial (RCT) as well (23). That trial observed a similar attack frequency at baseline and a similar response to verum injection as we did, but with a later and lower response in the placebo group. This lower response could be attributed to the addition of lidocaine in the verum injection, thereby increasing the possibility of placebo response and the usage of a lower dosage of verapamil, that was only initiated after one week. However, even with the later addition of verapamil and the lower verapamil dosage, groups began to converge after two weeks, highlighting the efficacy of verapamil. Thus, the main benefits of GON injection are, firstly, bridging the first few treatment weeks of a cluster period when patients still have many attacks and suffer severely because verapamil is not yet sufficiently effective and, secondly, lowering the required doses of verapamil in the first weeks of treatment, thereby reducing the risk of side effects. Despite the decrease in verapamil dose, the confidence interval for the difference is high (mean −60 mg; 95% CI = −4 to −116). The wide confidence interval probably reflects the individualised stepwise titration of verapamil, as the required dose can vary greatly between patients. This variability was reported in an open-label trial, where the mean verapamil dose for episodic CH patients was 354 mg, with a range of 240–600 mg (24). Furthermore, differences in the duration of effect of the GON injection further effects verapamil dosing, contributing to the broad confidence interval.
Close inspection of the individual verapamil dosage curves and attack frequencies shows that a verapamil dose increase was necessary in a subgroup of participants in the methylprednisolone group after the initial effect of the injection, which could be a reflection of the wearing-off effect of the GON injection. This is reflected in the later verapamil peak in the methylprednisolone group that can be observed in Figure 3. In this subgroup, it may be useful to administer a repeated GON-injection. However, safety and efficacy of repeated injections should be studied further. Available evidence, although limited, suggest this is safe (22).
Compared with two previous studies with GON injection (15,17), the absolute treatment effect was lower in our study, possibly because the previous studies were small (n = 16 and n = 24, respectively) and therefore overestimated the absolute treatment effect. However, the response rate to placebo was also lower, resulting in similar therapeutic gains compared with placebo. Moreover, the attack frequency at the start of our study was also lower, so the potentially achievable absolute treatment effect was also lower. This is probably due to the fact that in our study (i) participants were included earlier in their cluster episode (median of two weeks compared with four weeks (17–15) in earlier studies), when their attack frequency was still relatively low and (ii) prophylactic therapy with verapamil also started very early in the cluster period, immediately after GON injection. In all studies, the injection site was the same. Moreover, in this study, the historical duration of the cluster episodes was slightly longer in the verum group than in the placebo group, possibly leading to earlier spontaneous remission in the placebo group and a lesser absolute and statistical difference with the verum group.
Important strengths of our study include: (i) the large sample participants with ECH only and no chronic CH; (ii) the rapid inclusion of all participants at the beginning of a cluster episode before prophylactic treatment was started and before the cluster period had already ended spontaneously; (iii) the structured and detailed follow-up with an electronic attack journal and frequent telephonic consultations; and (iv) the investigator-initiated innovative add-on trial design that allowed the efficacy of GON injection with methylprednisolone to be studied in an ethical manner without causing unnecessary suffering to participants in the placebo group as explained in the methods section.
Some limitations and potential problems should also be discussed. First, we investigated whether GON injection with methylprednisolone as an add-on to a standardised titration protocol with verapamil would reduce the required dose of verapamil and whether improvement would start earlier with fewer side-effects. We did not directly compare GON injection (and placebo verapamil) with verapamil (and placebo GON injection) because, due to the known differences in the time course of efficacy (rapid onset but short-lasting for GON injection versus late onset but long-lasting for verapamil), this would not have been feasible and also not very clinically meaningful. Moreover, as explained earlier, we considered it unethical to treat participants with placebo alone without any other form of preventive treatment. However, this also gave us the opportunity to observe the effect of standard treatment with verapamil. This showed that the weekly attack frequency in both groups had decreased to a median of zero after four weeks.
Second, half of the participants received placebo GON injection with 2 ml of saline, which could theoretically have an indirect effect on the greater occipital nerve (GON) as was suggested in a meta-analysis of “control” injections (25). It has been suggested that the GON is compressed in some patients and that the volume of compound injected, whether verum or placebo, may decompress the GON and thus have a therapeutic effect. This theory has also been suggested in a study of high-volume injections around the GON (26). If placebo GON injection actually also had a therapeutic effect, it would have made demonstrating a statistical difference between GON injection with methylprednisolone and placebo more difficult.
Third, we did not exclude patients who had been previously treated with GON injection with methylprednisolone. However, we do not believe this led to bias. The number of participants who had had a previous GON injection did not differ significantly between the two treatment groups (n = 4 in the methylprednisolone group and n = 2 in the placebo group), nor did the treatment effect of a previous GON injection. Moreover, in a previous study, the effect of earlier GON injections did not appear to be predictive of the effect of later GON injections (22). Furthermore, it is impossible to predict that a newly diagnosed CH is not the beginning of a chronic CH. However, the probability of ECH is strongly in favour (85% vs. 15% prevalence). Moreover, we expect this to be balanced between verum and placebo.
Fourth, systemic corticosteroids are effective in CH. The effect of corticosteroid-containing GON injection is sometimes attributed to this systemic effect (27–28). Previous studies have used a combination of long-acting betamethasone (dipropionate 12.46 mg) and short-acting betamethasone (disodium phosphate 5.26 mg) (equivalent dose of 83 mg and 35 mg of oral prednisolone, respectively) or three injections of 3.75 mg of cortivazole each (equivalent dose of 62.5 mg of oral prednisolone), which are also unlikely to have a systemic effect (15,17).
Fifth and last, we used methylprednisolone, a relatively short-acting corticosteroid, without the addition of a local anaesthetic, as has been done in other studies (10,11,13,15,16). Although some think otherwise, there is no evidence for superiority of any other corticosteroid, whether long-acting or short-acting or a combination of both, or of addition of a local anaesthetic (14,15,16,17,29). Moreover, the addition of a local anaesthetic could have led to paraesthesia's and, therefore, possibly to unblinding.
In conclusion, a GON injection with 80 mg of methylprednisolone at the beginning of a cluster episode followed by standard therapy with verapamil did not show a reduction in verapamil use over the entire 12-week study period. However, it does provide faster improvement in attack frequency and intensity, lowers the required dose of verapamil and the risk of adverse events in the first four weeks than verapamil alone and is well tolerated and safe. We recommend using GON injection as a transitional treatment to overcome the delayed treatment effect of standard titration of verapamil in the first few weeks of a cluster period, when patients treated with low-dose verapamil alone still have many attacks and suffer severely because low doses of verapamil are not yet sufficiently effective or as a monotherapy in patient that have a contra-indication to verapamil.
Article highlights
GON injection with 80 mg of methylprednisolone at the beginning of a cluster episode followed by standard therapy with verapamil did not show a reduction in verapamil use over the entire 12-week study period. GON injection does provide faster improvement in attack frequency and intensity in the first four weeks than verapamil alone. GON injection lowers the required dose of verapamil and the risk of adverse events in the first four weeks. GON injection is well tolerated and safe. We recommend using GON injection as a transitional treatment to overcome the delayed treatment effect of standard titration of verapamil in the first few weeks of a cluster period.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251370324 - Supplemental material for Greater occipital nerve injection with methylprednisolone as transitional therapy in episodic cluster headache: Results from an RCT
Supplemental material, sj-docx-1-cep-10.1177_03331024251370324 for Greater occipital nerve injection with methylprednisolone as transitional therapy in episodic cluster headache: Results from an RCT by Roemer B. Brandt, Wim M. Mulleners, Emile Couturier, Johannes A. Carpay, Olivier H.H. Gerlach, Marieke Niesters, Joost Haan, Erik W. van Zwet, Michel D. Ferrari and Rolf Fronczek in Cephalalgia
Footnotes
Data availability
The data sets used and/or analysed during the present study are available from the corresponding author on reasonable request. The full trial protocol is available from the corresponding author on reasonable request.
Declaration of conflicting interests
WM reports Honoraria from Novartis, Teva, AbbVie, Lundbeck, Lilly and lecture fees from Lilly; EC reports consulting fees from Allergan, Amgen/Novartis, Lilly, Teva; JC reports consulting fees from Novartis, Lilly, Teva, Lundbeck, Pfizer; RF reports consulting fees from Novartis, Teva, AbbVie, Lundbeck, Lilly and lecture fees from Lilly. All other authors report no relevant conflicts of interest.
Ethical statement
Written informed consent was obtained from all participants according to the Declaration of Helsinki and the study protocol was approved by the ethical committee of the LUMC (METC-LDD.
Funding
This study was independently funded by de Hersenstichting (HA2017.01.03) and Innovatiefonds Zorgverzekeraars (3.649). The funders had no role in the study design, collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.