
Abstract
Migraine is a complex neurological disorder involving multiple neuropeptides that modulate nociceptive and sensory pathways. The most studied peptide is calcitonin gene-related peptide (CGRP), which is a well-established migraine trigger and therapeutic target. Recently, another peptide, pituitary adenylate cyclase-activating polypeptide (PACAP), has emerged as an alternative target for migraine therapeutics. This review compares the roles of PACAP and CGRP in preclinical mouse models of migraine. PACAP shares similarities with CGRP, and both are expressed in peripheral and central migraine-relevant regions. However, CGRP is more abundant in the trigeminal pain system, whereas PACAP is more prominent in parasympathetic ganglia that may contribute to autonomic aspects of migraine. PACAP and CGRP act on receptors that can activate overlapping but distinct intracellular signaling pathways. While both peptides elevate cAMP levels to activate protein kinase A, PACAP is more effective than CGRP at engaging an alternative cAMP pathway involving small G proteins, as well as Gq-mediated calcium pathways. Moreover, PACAP and CGRP induce similar migraine-like behaviors in mice, including cephalic and plantar mechanical allodynia, photophobia and non-evoked pain, but they do so by largely independent pathways. Notably, PACAP-mediated photophobia and mechanical allodynia symptoms are not blocked by CGRP-targeted therapies in mice. Finally, we discuss how preclinical PACAP and CGRP studies have translated to the clinic, with the exception of a PACAP type I receptor monoclonal antibody. Overall, CGRP and PACAP are likely to act by parallel and non-redundant roles in migraine pathophysiology, which suggests that a combined targeting of CGRP and PACAP may offer a more effective strategy for treating migraine.
This is a visual representation of the abstract.
Introduction
The discovery of calcitonin gene-related peptide (CGRP) as a key player in migraine has sparked research into other peptides that might also contribute to migraine (1,2). CGRP- and CGRP-receptor targeted therapies, including monoclonal antibodies, have been confirmed to be highly effective and safe, fueling further interest in peptide-driven migraine pathophysiology. However, there is a need for alternative treatments for patients who are not effectively treated by the CGRP-based drugs (2–5). One peptide has emerged as a therapeutic target for migraine: pituitary adenylate cyclase-activating polypeptide (PACAP) (6–9). PACAP is present as two isoforms of 38 and 27 amino acids, PACAP-38 and PACAP-27, respectively. Both can induce migraine in patients, which has driven research efforts to investigate its mechanisms and develop effective antagonists.
Peripheral and central distribution of PACAP and CGRP and their receptors
Peptide expression
CGRP and PACAP are both expressed in the meninges and peripheral ganglia involved in migraine pathophysiology (Figure 1) (10). Within the trigeminal nerve and ganglia, CGRP is far more abundant than PACAP. Indeed, while both peptides can be released from trigeminal ganglia neurons, PACAP is expressed in a relatively small subset of the neurons, approximately 20% in rats, compared to CGRP, which is found in 35–50% of neurons across species. Additionally, only approximately 9% of rat neurons co-express both neuropeptides (11,12). Furthermore, PACAP is poorly expressed in the trigeminal C-fibers found in the dura mater near the middle meningeal artery and in larger nerve branches (13). Notably, PACAP-38 is released by a distinct subset of trigeminal neurons that do not express CGRP, as well as by a subpopulation that co-expresses and co-releases both neuropeptides (11). By contrast, CGRP is richly expressed in the trigeminal ganglia and especially in the C-fibers (Figure 1) (1,11,13–16).
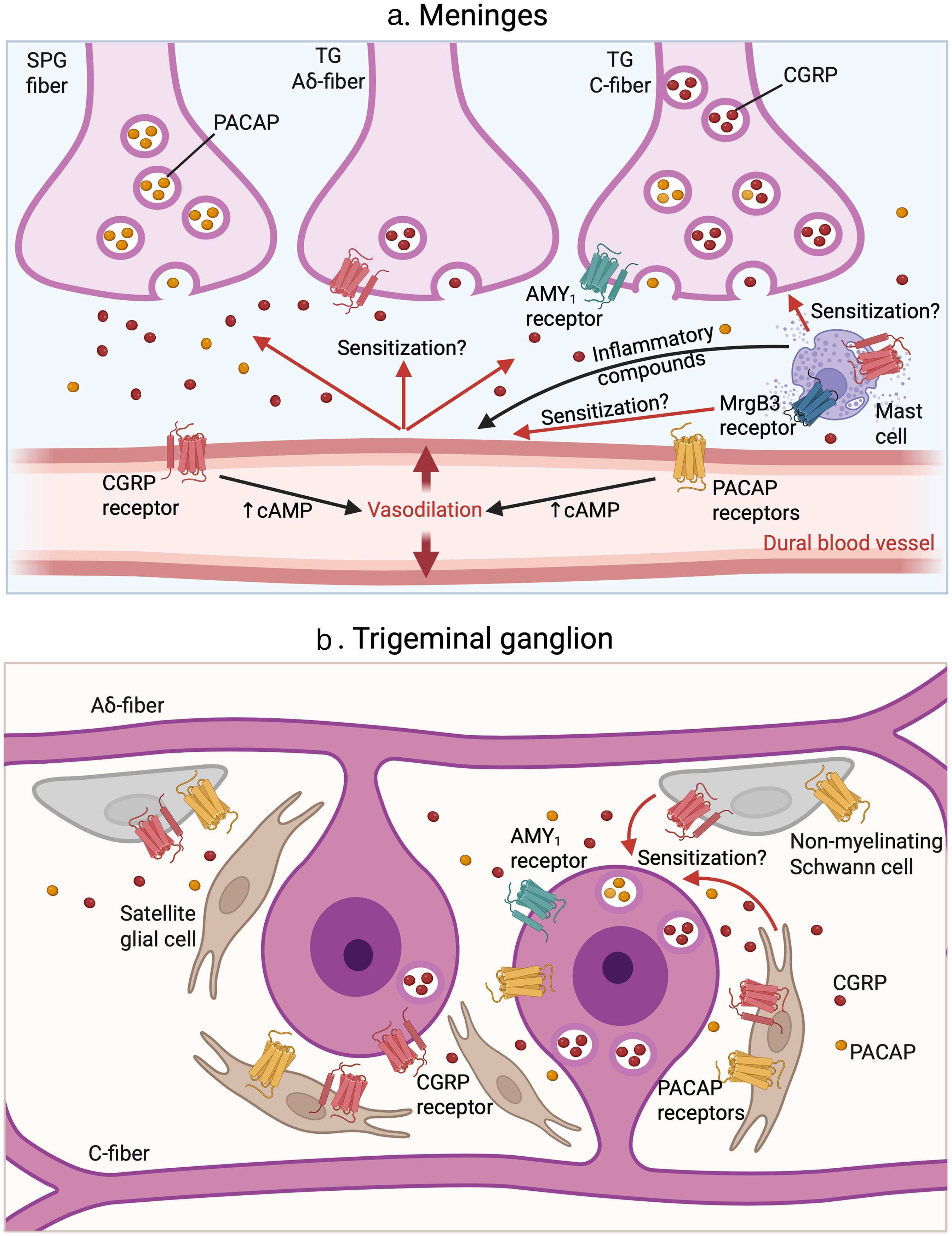
Pituitary adenylate cyclase-activating polypeptide (PACAP) and calcitonin gene-related peptide (CGRP) signaling in the trigeminovascular system. (a) At the meninges, perivascular nerve fibers in the dura from the sphenopalatine ganglia (SPG) and trigeminal ganglia (TG) (both C and Aδ fibers) release CGRP and PACAP. PACAP and CGRP can be co-released from the same C-fiber neurons, although PACAP levels are relatively low. The Aδ fibers release only small amounts of CGRP. The majority of PACAP is found in projections from the sphenopalatine ganglion (SPG). These peptides act on their respective receptors expressed on cranial blood vessels, mast cells, and the nerve fibers, as indicated. The canonical CGRP receptor is expressed on Aδ fibers and cranial blood vessels. The amylin 1 receptor (AMY1) receptor is expressed on C-fibers. Rodent mast cells express the CGRP receptor and the MrgB3 receptor, which mediates PACAP-induced degranulation. It is unknown whether PACAP receptors are expressed on meningeal nerve terminals. A generic PACAP receptor is represented to avoid the complexity of showing three different receptors (PACAP type I (PAC1), vasoactive intestinal polypeptide-PACAP 1 (VPAC1) and VPAC2). Mast cell degranulation could trigger further vasodilation via the release of inflammatory compounds. Vasodilation of blood vessels and mast cell degranulation may both contribute to the sensitization of the nerve terminal. This, in turn, further promotes the release of neuropeptides, perpetuating a vicious cycle of nociceptive signaling. (b) Within the trigeminal ganglion, cell bodies of unmyelinated C fibers release both PACAP and CGRP and express PACAP receptors and the AMY1 receptor, but not the canonical CGRP receptor. By contrast, the cell bodies of myelinated Aδ fibers release only minimal amounts of CGRP and express the canonical CGRP receptor. The PACAP and CGRP released from trigeminal neurons activate neighboring satellite glial cells and non-myelinating Schwann cells, which also express PACAP receptors and the canonical CGRP receptor. In response, these glial cells can contribute to the sensitization of trigeminal neurons, promoting a feed-forward loop of neuropeptide release that perpetuates migraine-associated signaling.
Regarding the expression of these neuropeptides in the satellite glia of the trigeminal ganglia, CGRP gene transcripts mostly encode procalcitonin (not CGRP), which may be involved in inflammatory responses (17), but its role, if any, in migraine is unknown. PACAP has been reported in a subset of satellite glial cells (15), although this is controversial. It was not seen by another group (18) and has been suggested to reflect binding to cell surface receptors rather than glial synthesis (19). Hence, the primary source of CGRP and PACAP in the trigeminal ganglia is from neurons.
Even though PACAP is less abundant in the trigeminal neurons, it is highly expressed in the parasympathetic sphenopalatine ganglia (18,20), from where it could trigger autonomic migraine symptoms (21–23). The presence of PACAP and CGRP in overlapping, but not identical groups of trigeminal neurons and the prevalence of PACAP in the sphenopalatine neurons raises the possibility that release of these neuropeptides is likely to be differentially regulated.
Receptor expression
Receptors for PACAP and CGRP are also expressed in the trigeminovascular system (Figure 1). Expression of canonical PACAP type I (PAC1) and also vasoactive intestinal polypeptide-PACAP 1 (VPAC1) and VPAC2 receptors within the small trigeminal ganglia neurons of humans and rodents is generally accepted (24,25). Additionally, PAC1, VPAC1 and VPAC2 were detected in rat sphenopalatine ganglion (19). Although it has been stated that PACAP receptors are expressed at both trigeminal and sphenopalatine ganglia nerve terminals at the dura (26), this remains to be fully established. In satellite glial cells, PAC1 and VPAC1 receptors have been detected (19). In mast cells, PACAP appears to act primarily through Mas-related G-protein coupled receptors. PACAP activation of the mouse Mrgprb2 receptor (27) and the rat MrgB3 (also known as Mrgprb3) receptor (28) is known to induce mast cell degranulation. Degranulation of dural mast cells releases a multitude of inflammatory and pro-inflammatory compounds that can sensitize neurons (Figure 1) (29).
PACAP is a potent vasodilator of cranial vessels, comparable to CGRP (Figure 1) (30). PACAP-induced vascular responses involve binding to PAC1, VPAC1 and VPAC2 receptors expressed on cranial vascular muscle cells, triggering cAMP-mediated signaling cascades (31,32). This activation ultimately opens ATP-sensitive and large conductance calcium-activated potassium channels, promoting potassium efflux. The resulting membrane hyperpolarization causes vasodilation. The process of potassium efflux and vasodilation has been proposed to sensitize perivascular neurons but remains debatable (33). In addition, PACAP may also induce vasodilation indirectly by activating mast cells in the meninges, potentially through the MRG family of receptors. However, this mechanism has also not yet been validated. Finally, PAC1 receptors have been identified in Schwann cells (34), and schwannoma cell lines have been shown to express both VPAC2 and PAC1 receptors (35), although the role these cells remains speculative.
CGRP receptors have been identified on meningeal blood vessels, subsets of neurons, satellite glia, mast cells and non-myelinating Schwann cells (13,36). CGRP canonical receptors, consisting of a G protein-coupled receptor, calcitonin receptor-like receptor (CLR), and a heterologous subunit, receptor activity-modifying protein (RAMP1), are located on Aδ fibers (37) and on the sphenopalatine ganglia (19). A second, non-canonical receptor called the amylin 1 (AMY1) receptor is likely present on C fibers (14,38). AMY1 consists of a related G protein-coupled receptor, calcitonin receptor (CTR) and RAMP1. CGRP receptors on vascular smooth muscle have long been known to cause vasodilation of blood vessels, including in the meninges (39). The CGRP receptors on satellite glia have been suggested to contribute to peripheral sensitization (1,14,36). In response to CGRP, satellite glia can release proinflammatory mediators that contribute to sensory neuron sensitization (40–42). CGRP promotes local neurogenic inflammation that includes degranulation of dural mast cells (29). While it is clear that rodent mast cells express the canonical CGRP receptor (CLR/RAMP1) and enable CGRP-induced degranulation (43,44), human mast cells have been reported to not express CGRP receptors (13). In addition, CGRP did not release histamine from human or porcine dural mast cells (45). However, a later study did report that primate mast cells express the CLR/RAMP1 receptor (46). Further studies are needed to determine whether human mast cells do express the CGRP receptor.
In summary, both CGRP and PACAP and their receptors are present within the trigeminovascular system and are both vasodilators. However, CGRP is more abundant and better characterized in terms of its release sites, receptor distribution and pro-nociceptive effects. PACAP, although less abundant within trigeminal nerves, acts through a broader set of receptors that may amplify mast cell degranulation. PACAP also plays a distinct role in autonomic pathways via parasympathetic ganglia. The activation of neurons, glia, mast cells and vasodilation is likely to contribute to a vicious cycle of nociceptive signaling that leads to peripheral sensitization, with additional neuropeptide release that sustains and amplifies migraine pain (47–49).
Beyond the ganglia and meninges, PACAP and CGRP and their receptors are broadly expressed across the central nervous system, including migraine-relevant brain regions such as the thalamus, cortex, cerebellum, brainstem nuclei and spinal cord (50–53). For transmission of nociceptive signals within the spinal cord, PACAP-38 was detected in numerous nerve fibers in the superficial layers of the dorsal horn of cervical, thoracic, lumbar and sacral segments, and a few fibers extended into the deeper layers of the spinal cord. In addition, PACAP-like immunoreactivity was seen in the intermediolateral cell column of the thoracic and sacral segments (53). The possible role of PACAP in the hypothalamus with respect to the control of sleep and circadian entrainment is particularly intriguing (54). CGRP and CGRP receptor expression in these and other central sites has recently been extensively reviewed (1).
Comparison of PACAP and CGRP receptors
Both PACAP and CGRP signal via G protein-coupled receptors (Figure 2). PACAP-38 acts through three canonical receptors, PAC1, VPAC1 and VPAC2 (55). CGRP primarily signals through two receptor complexes, CLR/RAMP1 (canonical) and CTR/RAMP1 (AMY1) (38). The RAMP1 subunit is required for cell surface expression of CLR and causes a conformation change in both CLR and CTR that allows CGRP binding. Therefore, CGRP and PACAP activate distinct, non-overlapping receptor systems, which have been previously defined as “parallel pathways” (56,57), meaning that these neuropeptides initiate separate signaling cascades that do not intersect at the receptor or early transduction level but may converge downstream at shared effectors involved in nociceptive processing.

Pituitary adenylate cyclase-activating polypeptide (PACAP) and calcitonin gene-related peptide (CGRP) intracellular signaling pathways. CGRP activates the canonical calcitonin receptor-like receptor (CLR)/receptor activity-modifying protein (RAMP1) receptor complex and the amylin 1 receptor (AMY1), which consists of calcitonin receptor (CTR)/RAMP1. PACAP acts on PACAP type I (PAC1), vasoactive intestinal polypeptide-PACAP 1 (VPAC1) and VPAC2 receptors, here represented by a single representative receptor. All these receptors signal via G protein-coupled pathways that stimulate cAMP production and activate protein kinase A. However, PACAP more strongly engages exchange protein directly activated by cAMP (Epac) after cAMP activation. Unlike CGRP, PACAP also preferentially activates phospholipase C (PLC) and inositol 14,5-triphosphate (IP3) signaling. Both peptides also promote receptor internalization and initiate β-arrestin-dependent endosomal signaling. Notably, endosomal signaling via the AMY1 receptor has not been demonstrated.
Interestingly, the VPAC1 and VPAC2 receptors can also associate with several RAMP subunits in heterologous expression systems and this association has been shown to bias the downstream signaling pathways (58–60). However, Ernstsen et al. (61) demonstrated that chronic administration of PACAP-38 induced mechanical allodynia in RAMP1 knockout mice that was comparable to that observed in wild-type mice. This indicates that PACAP receptors do not require the RAMP1 subunit, at least for this behavioral outcome.
Receptor complexity is heightened by the presence of splice variants in both PAC1 and CTR, which can alter downstream signaling (62). For example, alternative splicing of the PAC1 receptor produces variants with distinct ligand-binding and signaling properties, affecting their responsiveness to PACAP and vasoactive intestinal polypeptide and contributing to migraine pathophysiology (62). These variants can couple to different G proteins, primarily Gs and, in some cases, Gq, leading to divergent downstream effects such as cAMP-mediated vasodilation or phosphoinositide hydrolysis-induced vasoconstriction (24,63–66). The differences between PAC1 splice variants highlight the need for better pharmacological tools to dissect their roles in the trigeminovascular system.
Regarding CGRP receptors, unlike CLR, splicing isoforms of CTR show distinct structural and signaling properties that influence responses to CGRP, amylin, and antimigraine drugs (67–70). For example, the AMY1(Δ1–47) variant enhances cAMP signaling and reduces antagonist efficacy (67,71), whereas hCT(b) favors extracellular signal-regulated kinase (ERK)1/2 activation (68,72) and may drive nociception via G-protein-independent pathways (73,74). Given the limited data on their in vivo distribution, particularly in humans, studies using rodent models expressing humanized receptor splice variants are needed to clarify their roles in migraine pathophysiology and treatments.
PACAP and CGRP downstream signaling pathways
Most of the downstream intracellular signaling pathways are convergent for PACAP and CGRP with some differences (Figure 2). Indeed, like the CGRP pathway, activation of PACAP receptors stimulates adenylate cyclase, resulting in increased cAMP production (55). Although both recruit the canonical cAMP-protein kinase A signaling, compared to the CGRP receptors, PACAP receptors appear to preferentially recruit the non-canonical exchange protein directly activated by cAMP (Epac) pathway, which links cAMP to mitogen-activated protein kinase (MAPK) activation (75,76). Additionally, the PAC1 receptor presents biased agonism with PACAP-38 activating both cAMP and ERK pathways, while PACAP-27 activates only cAMP (77). Thus, while both CGRP and PACAP utilize cAMP, their downstream effectors can differ.
Both PACAP and CGRP receptor activation have the potential to additionally engage Gq-mediated calcium signaling pathways via phospholipase C and inositol 1,4,5-triphosphate (IP3) (78,79). However, the strength and significance of these couplings differ between the two systems. For CGRP receptors, coupling to Gq remains controversial, with mixed findings on calcium mobilization and IP3 production (80–82). By contrast, PACAP receptors show stronger evidence for effective Gq coupling (83). In standardized receptor models, the ability of CGRP to stimulate IP3 accumulation was over 200-fold weaker than its effect on cAMP (84,85), whereas PACAP-stimulated IP3 was only approximately 4–10-fold weaker than cAMP production (63), suggesting that Gq-mediated calcium signaling is more robust for PACAP than CGRP.
Additionally, CGRP signaling from endosomes via β-arrestin is well documented (24,86). This signaling is likely to be different for the two major CGRP receptors. The canonical CGRP receptor (CLR/RAMP1) internalizes rapidly and forms endosomal β-arrestin complexes, while the AMY1 receptor (CTR/RAMP1) remains longer at the cell surface (68,74). Importantly, endosomal signaling has been shown to be critical for CGRP-induced nociception (73,86). Similarly, the PAC1 receptor and both VPAC receptors also undergo β-arrestin-mediated internalization and may signal from endosomes (58,87), although the contribution of these internal pools to migraine pathophysiology remains largely unknown.
In summary, while PACAP and CGRP both utilize cAMP pathways, PACAP more effectively recruits additional signaling modules, including Epac, MAPK and calcium mobilization, across multiple cell types (Figure 2). These differences may underlie their distinct contributions to migraine pathophysiology and therapeutic efficacy.
Comparison of PACAP and CGRP migraine-like behavioral responses in mouse models
PACAP and CGRP both play roles in nociception and inflammation. However, although CGRP is recognized as a well-established pro-nociceptive and pro-inflammatory mediator (88), PACAP actions are less clear cut, with context-dependent modulatory effects on inflammation and nociceptive pathways. For example, intrathecal administration of PACAP-38 in mice (0.05–0.5 µg) produces a dose-dependent decrease in tail-flick latency, whereas higher doses (1–10 µg) evoke intense nocifensive behaviors such as biting, scratching, grooming, and licking of tail, paw and genital areas (53). A dual and modality-specific role for PACAP in pain modulation is suggested by studies with PACAP-deficient mice (89). These mice had reduced mechanical hyperalgesia and nocifensive responses, yet enhanced thermal allodynia. Complementing these findings, Akerman and Goadsby (90) demonstrated that neuronal PAC1 receptors mediate delayed activation and sensitization of second-order neurons in the trigeminocervical complex of rats, a key relay in migraine processing. This effect is inhibited by a PAC1 receptor antagonist, suggesting that PACAP-38 promotes central sensitization through PAC1 receptor activation (90). Importantly, neither PACAP nor CGRP induce significant motor impairment at doses that trigger migraine-like symptoms (61,91,92), which ensures that observed sensory and pain behaviors represent nociceptive effects rather than motor deficits.
Overall, although the distribution of PACAP, CGRP and their receptors is largely conserved across species, as reviewed previously (1,10), some differences exist that may affect translational relevance. For example, PAC1 splice variants such as PAC1hiphop and PAC1s are characterized in rodents but not confirmed in humans, potentially altering receptor signaling and drug responses (62). Pharmacodynamic differences also exist; for example, CGRP receptor antagonists such as telcagepant show species-specific binding and efficacy profiles (93). These discrepancies highlight species-specific limitations in translating rodent data to humans and underline the importance of using human tissues or humanized mouse models to improve clinical relevance.
The most commonly used behavioral readout has been cephalic and plantar mechanical allodynia, characterized by heightened sensitivity to normally non-painful tactile stimuli around the head and facial or hindpaw areas, respectively (94). Both are robustly induced by PACAP and CGRP administration. The current preclinical literature on PACAP and CGRP effects on migraine-like symptoms in mouse models are described below and summarized in Table 1.
Pituitary adenylate cyclase-activating polypeptide (PACAP) vs. calcitonin gene-related peptide (CGRP) in migraine-like behavioral symptoms in mouse models.
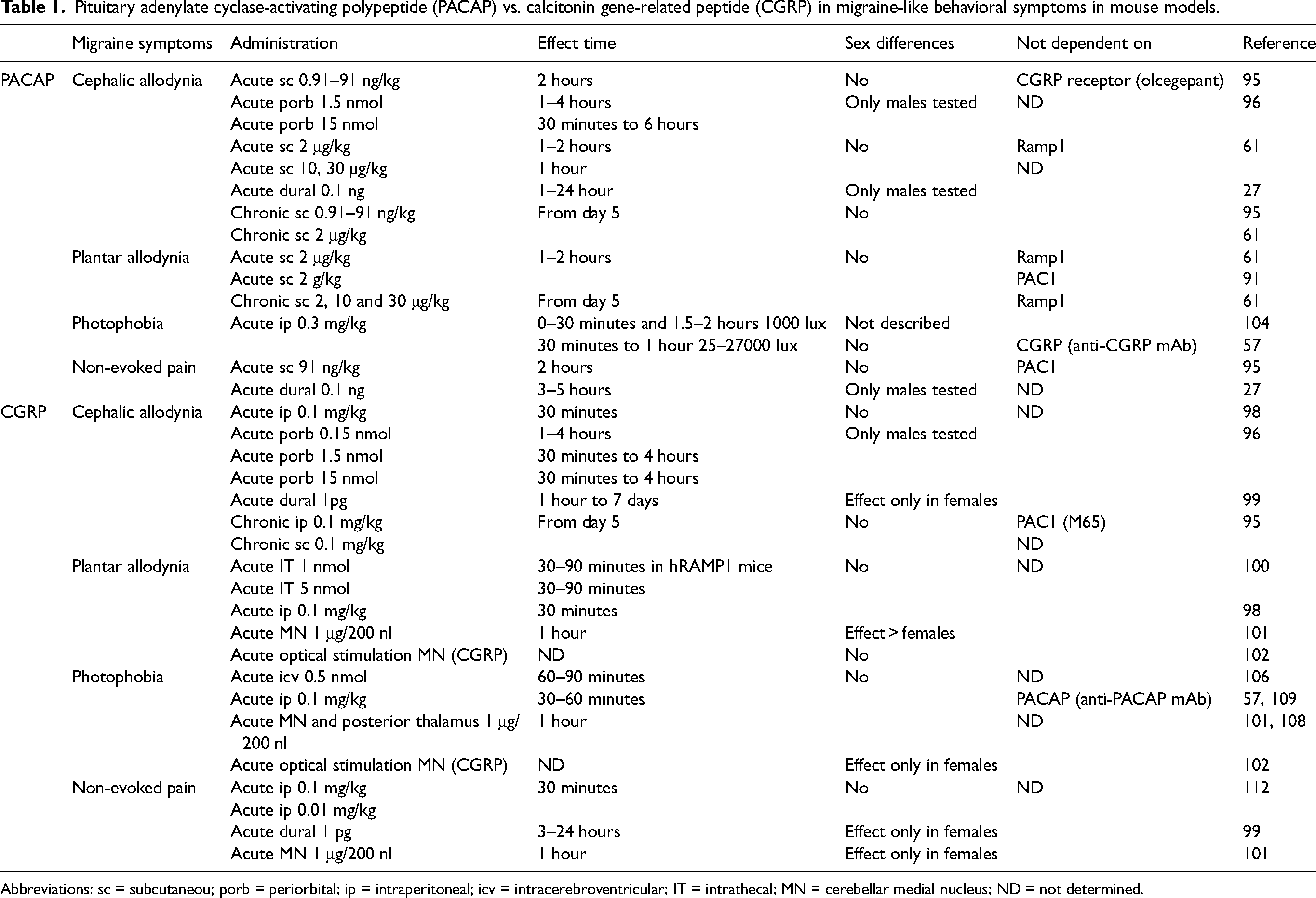
Abbreviations: sc = subcutaneou; porb = periorbital; ip = intraperitoneal; icv = intracerebroventricular; IT = intrathecal; MN = cerebellar medial nucleus; ND = not determined.
Mechanical cephalic allodynia
PACAP induces mechanical cephalic allodynia in mice of both sexes following subcutaneous injection (61,95), although studies using periorbital or dural administration have so far been conducted only in males (27,96). PACAP-induced periorbital allodynia is notably prolonged. PACAP-38 injection into the mouse dura mater produced a sustained increase in periorbital allodynia lasting up to 24 hours, which was dependent on the MrgprB2 receptor (27). PACAP-induced periorbital allodynia was unaffected by CGRP-blocking antibodies, suggesting independent pathways (61). Indeed, chronic PACAP administration, similar to CGRP, induces periorbital sensitization starting from day 5. This sensitization is not reversed by the CGRP receptor antagonist olcegepant (95), consistent with PACAP and CGRP mediating allodynia by distinct and independent mechanisms. In addition to pharmacological studies, Ernstsen et al. (61) also used a genetic approach with mice lacking the RAMP1 subunit. RAMP1 is an obligate subunit for the CGRP receptor (97), and so, as a control, they showed that RAMP1 knockout mice did not respond to CGRP. By contrast, there was no difference in PACAP induced mechanical periorbital allodynia between RAMP1 knockout and wild-type mice, again indicating independent pathways for PACAP and CGRP (61).
CGRP induces mechanical cephalic allodynia in mice regardless of the sex with intraperitoneal (98), subcutaneous (95) and periorbital injections (96). However, dural (but not intraplantar) administration of CGRP in both mice and rats yielded mechanical cephalic allodynia only in females (99), which contrasts with PACAP causing this allodynia in stress-primed males (27). The periorbital CGRP-induced mechanical cephalic allodynia was shown to be fully blocked by anti-CGRP receptor antagonists, confirming its major dependency on the CGRP receptor (CLR/RAMP1) (96). As with PACAP, chronic administration of CGRP given every other day induced mechanical cephalic allodynia beginning on day 5. Notably, this sensitization was not significantly attenuated by the PAC1 receptor antagonist M65 (95), meaning that this CGRP-induced periorbital allodynia predominantly occurs through a PACAP-independent pathway.
Nonetheless, these data do not exclude some potential intersection between the two pathways. For example, Jansen-Olesen et al. (15) reported a concentration-dependent release of CGRP by PACAP-38 in the trigeminal nucleus caudalis but not in the trigeminal ganglia. Consequently, it is possible that there is convergence between PACAP and CGRP pathways at the level of second-order neurons leading to central sensitization.
Mechanical plantar allodynia
Subcutaneous PACAP administration in rodents induces acute hindpaw plantar responses. Mechanical plantar allodynia is increased one to two hours after subcutaneous PACAP-38 administration compared to vehicle-treated controls. As with periorbital allodynia, no significant differences in mechanical plantar allodynia were observed between RAMP1 knockout mice and their wild-type littermates (61). PACAP acts independently of both the CGRP receptor and the CGRP peptide, as indicated by the lack of differences between mice treated with PACAP-38 and a CGRP monoclonal or a control antibody (61). The relevant receptors have been investigated by Guo et al. (91), who showed that both VPAC1 knockout and VPAC2 knockout mice had reduced PACAP38-induced mechanical hindpaw allodynic responses compared to wild-type mice, suggesting these receptors collectively contribute to this hypersensitivity. By contrast, the PACAP-38 induced hindpaw hypersensitivity in PAC1 knockout mice did not differ from wild-type (91). Therefore, PACAP-38 likely mediates mechanical plantar allodynia in mice through combined actions on VPAC1 and VPAC2 receptors.
Intrathecal (100) and intraperitoneal (98) CGRP and subcutaneous PACAP (61) induce hindpaw hypersensitivity in the plantar area. Intrathecal injection of 1 nmol CGRP significantly increased paw withdrawal frequency in nestin/hRAMP1 mice that have elevated expression of the hRAMP1 subunit of the CGRP receptor (100). By contrast, control mice showed no significant response at this dose and required a higher dose of 5 nmol CGRP to trigger hindpaw hypersensitivity. This difference suggests that RAMP1 expression is functionally rate-limiting for CGRP-induced mechanical nociception. By contrast to intrathecal CGRP administration, peripheral intraplantar and subcutaneous injection of CGRP in the subscapular region did not alter the mechanical withdrawal threshold (100). Subsequent studies demonstrated that either central CGRP administration into the cerebellar medial nuclei (MN) or optogenetic stimulation of CGRP-expressing MN neurons (MNCGRP) induced plantar mechanical hypersensitivity (101,102). This suggests that MNCGRP neurons play a role in inducing migraine-like behaviors and may prove to be therapeutic targets. The pharmacological approach produced a stronger response in female mice, whereas optogenetic stimulation induced comparable hypersensitivity in both sexes. This suggests sex-dependent differences in the response to exogenous CGRP, which likely activates CGRP receptor-expressing neurons in the MN, but not the neurons expressing the CGRP peptide (101,102). Additionally, peripheral (intraperitoneal) injection of CGRP induces plantar hypersensitivity in CD1 mice (98).
Photophobia
Another common migraine symptom is photophobia, consisting of aversion to light (103). Markovics et al. (104) reported that intraperitoneal PACAP-38 administration induced photophobia and meningeal vasodilation in wild-type CD1 mice but, surprisingly, not in PACAP knockout mice. The reason for lack of an effect in PACAP knockout mice is not known, although Markovics et al. (104) speculated that exogenous PACAP acted by triggering release of endogenous PACAP in the trigeminal nucleus caudalis, which is inside the blood–brain barrier and presumably would not be exposed to exogenous PACAP. This endogenous release would not occur in the knockout mice. Indeed, whether PACAP acts centrally or peripherally remains an open question and, while speculative, this result suggests that both sites may be involved. PACAP knockout mice also exhibit significantly reduced nitroglycerin-induced light aversion compared to wild-type, suggesting that PACAP has a crucial role in mediating the photophobic migraine-related behavior (104). However, it is important to acknowledge that PACAP knockout mice have been reported to exhibit altered behavioral phenotypes, including hyperactivity, sudden jumping episodes in open field tests, increased exploratory drive and reduced anxiety-like behavior in paradigms such as the elevated plus maze, emergence and novel object tests. These baseline behavioral differences could potentially confound the interpretation of light-aversion outcomes in these models (105). Nevertheless, PACAP-induced light aversion was later confirmed by Kuburas et al. (57), who showed that PACAP induced light sensitivity in CD1 wild-type mice. Unexpectedly, the response to PACAP was not seen in about one-third of the outbred CD1 mice. This resilience to PACAP was a stable and inheritable trait and differed from CGRP, which induced light aversion in almost all mice. Notably, PACAP-induced photophobia was not blocked by an anti-CGRP antibody, and CGRP-induced photophobia was not blocked by an anti-PACAP antibody, indicating that PACAP and CGRP mediate photophobia via parallel, non-redundant pathways (57).
Similar to PACAP, CGRP injections have also been shown to induce light aversion in mice. Light aversion in dim light was observed by Russo et al. (106) following intracerebroventricular administration of CGRP in transgenic nestin/hRAMP1 that overexpress the RAMP1 subunit of the CGRP receptor in neurons. By contrast, intracerebroventricular CGRP injection into wild-type mice only caused light aversion when the mice were exposed to bright light and exploratory drive was reduced by pre-exposure to the testing chamber (107). Light aversion was seen in both sexes. Subsequent studies indicate that CGRP is likely to act at multiple sites in the brain. Injection of CGRP directly into the posterior thalamus and cerebellar MN induced light aversion to dim light in both sexes (101,108). By contrast, injection of CGRP into the hippocampus did not cause light aversion (108). Similarly, optogenetic stimulation of MNCGRP neurons increased light aversion, but selectively in female mice (102). Finally, it was shown that peripheral (intraperitoneal) injection of CGRP could also induce light aversive behavior in both CD1 and C57BL/6J wild-type mice that was similar to that induced by central administration (109). A genetic approach using the nestin/hRAMP1 sensitized mice revealed that, unlike for centrally injected CGRP, neuronal CGRP receptors were not the rate-limiting factor for peripheral CGRP actions (57,109). The rate-limiting site of peripheral CGRP receptors required for photophobia is not known. Nonetheless, the ability of both central and peripheral CGRP to cause photophobia in mice is reminiscent of the suggestion by Markovics et al. (104) that PACAP can also act both centrally and peripherally to cause photophobia.
Non-evoked pain
Non-evoked pain is one of the most frequently assessed migraine-related symptoms and, in mouse models, it is commonly measured using the mouse grimace scale (110,111). Subcutaneous PACAP injections led to an increase in grimace scores two hours post-injection regardless of the sex (95). PACAP-38 injection into the dura mater of stress-primed mice produced a sustained increase in grimace scores of male mice lasting up to five hours that was dependent on the MrgprB2 receptor (27).
CGRP has been shown to induce non-evoked pain following peripheral or central administration. As assessed by the mouse grimace scale and automated squint analysis, CGRP triggers non-evoked pain when administered via dural injection (99) or intraperitoneally (112–114). This was abolished by pretreatment with a CGRP monoclonal antibody (112). Interestingly, a central injection of CGRP into the MN of the cerebellum induced squinting behavior specifically in female mice (101). By contrast, optical stimulation of MNCGRP neurons did not produce a significant increase in squinting (102). These differences reflect the activation of distinct neuronal populations that express either the CGRP ligand or its receptor.
Translation of preclinical studies targeting PACAP and CGRP to clinical trials
Preclinical studies have played a pivotal role in shaping clinical trials targeting PACAP and CGRP pathways in migraine. The migraine treatments targeting CGRP and PACAP pathways are summarized in Figure 3.

Pituitary adenylate cyclase-activating polypeptide (PACAP) and calcitonin gene-related peptide (CGRP) targeted migraine therapeutics. Food and Drug administration-approved drugs targeting (a) PACAP or (b) CGRP pathways are boxed in green, discontinued compounds are shown in red and those undergoing testing are shown in purple. The drugs in purple that are still at a preclinical testing phase have a mouse represented next to their name. All other compounds have undergone or are currently undergoing clinical trials. The dotted lines indicate that gepants and erenumab are less effective at amylin 1 receptor (AMY1) receptor. PACAP and CGRP receptors are shown on a generic cell to represent the cascade of signaling that leads to migraine pathophysiology. PAC1 = PACAP type I; VPAC = vasoactive intestinal polypeptide-PACAP.
Animal models revealed that PACAP can induce migraine-like behaviors, such as photophobia and mechanical cephalic allodynia, and that these behaviors could be attenuated by inhibitors of PACAP signaling, including a PACAP monoclonal antibody and a PAC1 antagonist (57,95). The efficacy of the PACAP monoclonal antibody Lu AG09222 in a phase 2 clinical trial is consistent with these preclinical insights (115). It should be noted that, although a clinical trial with another PACAP monoclonal antibody (LY3451838) was discontinued, this appears to have been primarily a result of recruitment issues that precluded a decision on efficacy (116). Indeed, given the demonstrated heritable responsiveness to PACAP for light-aversion in mouse preclinical models (57), it is possible that the LY3451838 clinical trial did not enroll enough PACAP-responsive individuals to detect a meaningful therapeutic effect of the antibody. Importantly, as discussed above, these preclinical PACAP actions were shown to be independent of CGRP signaling by using a CGRP receptor antagonist or CGRP monoclonal antibodies (57,109). This preclinical independence of CGRP and PACAP actions was also demonstrated in a clinical study showing that PACAP-38 can still trigger migraine attacks in patients treated with a CGRP monoclonal antibody, eptinezumab (117). This translational consistency underscores the importance of preclinical models for further developing PACAP-targeted therapies.
By contrast to translation of studies that directly target the PACAP ligand, studies that target the PACAP receptors have not yet yielded a clear connection between rodents and humans. A PAC1 receptor antagonist M65 effectively reversed chronic mechanical periorbital allodynia and blocked enhanced cortical spreading depression in a mouse model of opioid-exacerbated migraine (118). These findings suggest that agents such as M65 could offer therapeutic benefit for specific conditions such as opioid-induced hyperalgesia and medication overuse headache, but it has not yet been clinically tested. A PAC1 antibody, Ab181, showed promise in reducing central nociception in rats (119), but was not advanced to human trials. Another PAC1 antibody, AMG 301, failed in a phase 2 clinical trial, although the reasons were not clear (e.g. lack of target engagement, receptor redundancy, recruited population, etc.) (120). In this context, it is important to consider species-specific differences in PAC1 receptor splicing variants. The relevance of specific splice variants to migraine pathophysiology may vary across individuals, and the variants targeted in rodents may not fully correspond to those driving migraine attacks in humans. Additionally, receptor localization may significantly influence therapeutic outcomes. Distinct receptor pools, such as those at the plasma membrane versus within endosomal compartments, can mediate different signaling cascades. This has direct implications for drug design: monoclonal antibodies typically act on cell surface receptors, whereas small molecules, depending on their chemical properties and tailored features, may access intracellular compartments and modulate endosomal receptor signaling. As such, receptor localization must be considered when developing new therapeutic agents and interpreting the efficacy of existing ones. Thus, for PAC1, preclinical efficacies have not yet led to clinical validation.
Meanwhile, CGRP-targeted therapies have successfully transitioned from bench to bedside. The CGRP receptor antagonists (gepants) rimegepant, ubrogepant, atogepant and zavegepant (121), as well as the monoclonal antibodies erenumab, eptinezumab, fremanezumab and galcanezumab (122), have demonstrated clinical efficacy for both acute and preventive treatment of migraine. This success has validated the translational strength of preclinical CGRP research. Only a couple of early developed gepants were discontinued in the translation from animal models to patients. For example, olcegepant demonstrated clinical efficacy but never reached commercialization as a result of its inability to be administered orally, although it continues to be widely used in preclinical studies. Telcagepant was also one of the earliest gepants developed but it was discontinued because of liver toxicity in long-term use. Interestingly, several gepants and erenumab can also block CGRP actions at AMY1 receptors to some degree in preclinical tests, although the clinical relevance remains speculative (123). A specific amylin and calcitonin receptor antagonist, AC187, has been used in animal models but not yet considered for clinical trials (38). Taken together, these lines of investigation highlight how mechanistic insights from animal models continue to inform and refine therapeutic strategies for migraine.
Conclusions and future directions
In conclusion, although CGRP remains the most extensively studied and therapeutically validated neuropeptide in migraine pathophysiology, growing preclinical and clinical evidence supports PACAP as a parallel, non-redundant trigger of migraine. Both peptides induce overlapping migraine-like symptoms in mouse models, including mechanical periorbital and plantar allodynia, photophobia and non-evoked pain, yet act through distinct receptor systems and intracellular signaling pathways. CGRP predominantly signals through the CLR/RAMP1 and possibly the AMY1 receptors, and both can be targeted to varying degrees by gepants and monoclonal antibodies. By contrast, PACAP engages multiple receptors (PAC1, VPAC1, VPAC2 and MRGPRX2/B3/B2) and recruits additional pathways such as Epac/MAPK and Gq-calcium signaling. While the actions of CGRP in trigeminal sensory neurons and satellite glia are well defined, the functional roles of PACAP in trigeminal and parasympathetic circuits remain largely underexplored. Despite early PAC1-based approaches being unsuccessful, promising results with ligand-targeting antibodies, such as Lu AG09222, suggest a path forward. As a future perspective, combined CGRP and PACAP targeted therapies should be considered. Indeed, there is an on-going clinical trial combining the PACAP antibody Lu AG09222 with ubrogepant (124). A strategy targeting multiple neuropeptides could offer improved treatment for migraine.
Article highlights
Preclinical and clinical evidence support PACAP and CGRP as parallel, non-redundant triggers for migraine.
A strategy targeting multiple neuropeptides could offer improved treatment for migraine.
Footnotes
Acknowledgments
Figures created with Biorender.com
Declaration of conflicting interests
AFR is a consultant for Lundbeck, Abbvie, Pfizer, Vedana Therapeutics, Arrowhead Pharmaceuticals, Omeros Corporation and Delphian Therapeutics, and holds patents on the CGRP monoclonal antibody for photophobia and diarrhea, on the PACAP monoclonal antibody for photophobia and on the CGRP HO enhancer element. ADP is a consultant for Delphian Therapeutics and AK is a consultant for Vedana Therapeutics. The contents do not represent the views of the US Department of Veterans Affairs or the United States Government.
Funding
This work was supported by a research grant from the National Institutes of Health R01NS129573 (AFR).