
Abstract
Aim
Meningeal nociceptors within the trigeminal ganglion are important contributors to migraine pathogenesis because they transmit pain signals from the dura mater to the central nervous system. As such, pharmacological interventions that target these peripheral neurons might offer new avenues for migraine treatment. In this context, ATP-sensitive potassium (KATP) channels have garnered increasing attention as potential modulators of meningeal nociception. Human experimental studies support this hypothesis, showing that intravenous infusion of levcromakalim, a KATP channel opener, induces migraine attacks in people with migraine and mild, transient headache in healthy adults. However, the precise anatomical site and mechanism of action remain incompletely understood.
Methods
To address these gaps, we conducted in vivo single-unit electrophysiological recordings of 36 meningeal nociceptors (23 Aδ- and 13 C-fibers) in the trigeminal ganglion of anesthetized male and female rats. We measured spontaneous firing rates before and up to four hours after a 20-minute continuous intracarotid infusion of levcromakalim (1.43 mg/kg or 0.14 mg/kg) or vehicle (71.4% ethanol).
Results
Levcromakalim at 1.43 mg/kg activated nine (69%) of 13 nociceptors, compared with one (11%) of nine in the vehicle group (p = 0.012). Activation rates did not differ between Aδ-fibers (6 of 8, 69%) and C-fibers (3 of 6; 50%; p = 1.00), or between male (6 of 8; 75%) and female animals (3 of 5; 60%; p = 0.61). Moreover, levcromakalim at 0.14 mg/kg activated only three (21%) of 14 nociceptors.
Conclusions
Taken together, our findings demonstrate that KATP channel opening mediates activation of meningeal nociceptors, providing a mechanistic basis for previous observations in humans. The development of KATP channel blockers might therefore hold therapeutic promise for migraine by inhibiting meningeal nociceptors.
This is a visual representation of the abstract.
Introduction
Migraine is a disabling neurological disorder that afflicts more than one billion people worldwide. 1 The clinical presentation is characterized by recurrent attacks of moderate to severe headache, often accompanied by photophobia, phonophobia, nausea and vomiting. 2 Although the neurobiological underpinnings of migraine remain incompletely understood, it is widely accepted to involve the trigeminal nerve and its axonal projections to the meninges and its blood vessels (i.e. the trigeminovascular system).3,4 These axonal fibers contain certain neuropeptides, including calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating polypeptide (PACAP), which are released in response to stimulation. 3 Notably, human experimental studies have documented that intravenous infusion of CGRP or PACAP induces migraine attacks in people with migraine and causes mild, transient headache in healthy adults.5,6
In search of more effective therapies, focus is starting to shift from targeting individual neuropeptides to examining their convergent downstream signaling pathways.7,8 Accumulating evidence suggests that peptidergic migraine triggers converge on cAMP-dependent signaling, ultimately causing the opening of specific potassium channels.7,9 Among these, ATP-sensitive potassium (KATP) channels have emerged as important contributors to migraine pathogenesis.7,10 Indeed, intravenous infusion of levcromakalim, a KATP channel opener, potently induces migraine attacks in people with migraine, while eliciting no more than mild headache in healthy adults.7,11 Furthermore, levcromakalim triggers cluster headache attacks in individuals with cluster headache and migraine-like headache in those with post-traumatic headache.12,13 Consistent with these clinical findings, levcromakalim also elicits nocifensive behavior in animal models of migraine, modifying animal behavioral readouts potentially related to migraine.14–16 These findings suggest that KATP channel opening is a pathogenic contributor across both primary and secondary headache disorders. Thus, blocking these channels holds the promise of greater therapeutic efficacy by inhibiting the downstream action of multiple peptidergic migraine triggers, rather than focusing on a single neuropeptide.
Despite important insights from human experimental studies, several knowledge gaps remain; foremost among them is the precise anatomical site and mechanism by which pharmacological KATP channel opening induces migraine. In most instances, KATP channel opening hyperpolarizes cells, including neurons and vascular smooth muscle cells (VSMCs), which is paradoxical given levcromakalim's ability to elicit migraine attacks.7,17–19 Furthermore, preclinical evidence has found that central administration of levcromakalim exerts anti-nociceptive effects,20–26 whereas oral ingestion of a neuronal KATP channel opener failed to induce migraine attacks in people with migraine. 27 A compelling hypothesis reconciles previous findings and suggests that opening of KATP on meningeal VSMC causes potassium efflux, which disrupts local potassium gradients and discharges perivascular afferents innervating the dura. This might initiate ascending trigeminal pain signals that eventually lead to migraine headache. However, this proposal depends on the critical question of whether KATP channel opening ultimately activates meningeal nociceptors.
To address this gap, we sought to determine whether intracarotid administration of levcromakalim activates mechanosensitive meningeal nociceptors, specifically examining both unmyelinated C-fibers and thinly myelinated Aδ-fibers. We used in vivo single-unit electrophysiological recordings in anesthetized male and female rats, closely mirroring human migraine provocation models to maximize the translational relevance of our findings.
Methods
All experiments were approved by the animal care committees of Beth Israel Deaconess Medical Center and Harvard Medical School. The study complied with the guidelines of the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Study design
The primary aim of the study was to evaluate whether levcromakalim activated meningeal nociceptors in rats. This was investigated through 5-hour electrophysiological single-unit recordings (including one hour of baseline and four hours after start of infusion), after infusion of one of two doses of levcromakalim or vehicle.
Surgical preparation
Male and female Sprague–Dawley rats (199–347 g; Taconic, Germantown, NY, USA) were anesthetized with urethane (1.5 g/kg intraperitoneally) and received atropine (0.4 ml subcutaneously) to reduce oral secretions. We then cannulated the femoral vein and common carotid artery for drug infusion. We opted for intracarotid infusion for its direct delivery to cephalic sites relevant to migraine, and because human studies have most aptly measured biochemical changes during migraine attacks in the jugular/cervical vasculature. Core temperature was maintained at 37°C using a feedback-controlled heating pad. We delivered O2 via arterial ventilation using an endotracheal tube, ensuring pO2 > 98%.
Following this, rats were paralyzed with rocuronium bromide (10 mg/ml and 1 ml/h continuous intravenous infusion). A craniotomy was performed above the left transverse sinus for electrical and mechanical stimulation of the dura mater, which was kept moist with isotonic saline. A second craniotomy was conducted from approximately 1–4 mm caudal to 1–4 mm lateral (right side) to the bregma, and the dura mater was removed to permit neural recording from the left trigeminal ganglion. A microelectrode was then advanced at an oblique angle to penetrate the cortex and enable recordings from the contralateral trigeminal ganglion. Figure 1 provides an overview of the experimental set-up.

Experimental set-up for stimulation and recording.
Neural recording
We used platinum-coated tungsten microelectrodes (150–300 kΩ impedance) to record single-unit activity of trigeminal ganglion neurons. Meningeal nociceptors were identified by their constant latency response to electrical stimulation of the dura mater (0.5 ms pulse at 5 mA, 0.75 Hz) delivered via a bipolar electrode. We classified neurons as Aδ-fibers (conduction velocity >5 m/s) or C-fibers (1.5–5 m/s), assuming a conduction distance of 12 mm between the dura mater and the trigeminal ganglion. Spike acquisition and waveform discrimination were performed using Spike 2 software (CED, Cambridge, UK), which enabled sorting and analysis of neural activity. Neurons were eligible for analysis if they exhibited a clearly identifiable mechanoreceptive field on the dura mater. This was confirmed by consistent spike firing in response to mechanical probing with blunt forceps or von Frey filaments. To ensure physiological relevance, only neurons exhibiting spontaneous firing during the baseline period were eligible for further analysis.
Experimental paradigm
The spontaneous neuronal firing rate was continuously recorded throughout the entire experimental session to capture baseline activity and drug-induced alterations. Following a stable baseline period, intracarotid infusion of either one concentration of levcromakalim or ethanol (71.4%) was administered. The volume administered was the same (1 ml/kg) for all concentrations of levcromakalim and the vehicle.
Levcromakalim was administered at two concentrations: 1.43 mg/kg, dissolved in 71.4% ethanol, and a 10-fold lower, human-equivalent dose of 0.14 mg/kg in 7.1% ethanol. Dose selection followed established human-to-animal conversion guidelines and was consistent with concentrations used in previous studies.14,15,28,29 Each dose was delivered via continuous intracarotid infusion over a 20-minute period. The 20-minute infusion was chosen over bolus administration to align our translational approach with human models of migraine, where participants receive continuous infusion over 20 minutes.
We likewise recorded heart rate immediately prior to infusion (baseline) and every 10 minutes for 60 minutes after infusion of either levcromakalim or vehicle, using a rat pulsoximeter (PhysioSuite; Kent Scientific, Torrington, CT, USA).
Data analysis
Baseline was defined as a minimum 30-min period during which neuronal firing remained stable. A neuron was considered activated when its firing rate exceeded 2 standard deviations (SDs) above the baseline mean for at least 10 consecutive minutes. Activation duration was calculated as the total number of minutes in which firing rates, averaged in 60-second bins, remained above this threshold. We also quantified the number of spikes that surpassed the activation threshold over the entire recording. Response latency was defined as the time between the start of levcromakalim or vehicle infusion and the onset of neuronal activation.
The primary outcome was the number of activated neurons in response to levcromakalim compared with vehicle, analyzed using Fisher's exact test. Additional analyses assessed the impact of sex (male vs. female) and fiber type (Aδ- vs. C-fiber) using Fisher's exact test.
Time to activation and magnitude of activation were compared using either the Wilcoxon rank sum test or Student's t-test, based on the distribution of data. Normality was assessed using quantile-quantile plots and visual inspection. Data conforming to normal distribution are presented as the mean ± SD, whereas non-normally distributed data are reported as the median with the interquartile range (IQR).
We performed sample size calculation for the comparisons between levcromakalim and vehicle based on an expected 60% difference in activation rate. Sample size calculation was performed with 80% power and a one-sided 5% alpha. We relied on unequal group sizes, with a ratio of 1 (vehicle) to 1.5 (levcromakalim), to facilitate additional comparisons between Aδ and C-fibers, as well as male and female animals. This estimated sample sizes of nine for the vehicle group and 13 for the levcromakalim groups.
To assess longitudinal changes in firing rate, Friedman's test was used across repeated measures. Post-hoc comparisons between baseline and each hour following infusion (first through fourth hour) were conducted using the Wilcoxon signed rank test. p < 0.05 was considered statistically significant. No corrections were made for multiple comparisons. All statistical analyses were performed using R statistical software, version 4.2.0 (R Foundation, Vienna, Austria).
Results
Neurons
Single-unit recordings were obtained from 36 meningeal nociceptors within the trigeminal ganglion. These included 23 Aδ-fibers and 13 C-fibers, each confirmed by their responses to electrical and mechanical stimulation of the dura mater over the ipsilateral transverse sinus.
Intracarotid levcromakalim (1.43 mg/kg) was tested in 13 neurons (8 Aδ, 5 C fibers; 7 male, 6 female) and at 0.14 mg/kg in 14 neurons (8 Aδ, 6 C fibers; 9 male, 5 female). A vehicle control (71.4% ethanol) was tested in nine neurons (6 Aδ, 3 C fibers; 4 male, 5 female). Examples of neuronal recordings are presented in Figure 2.
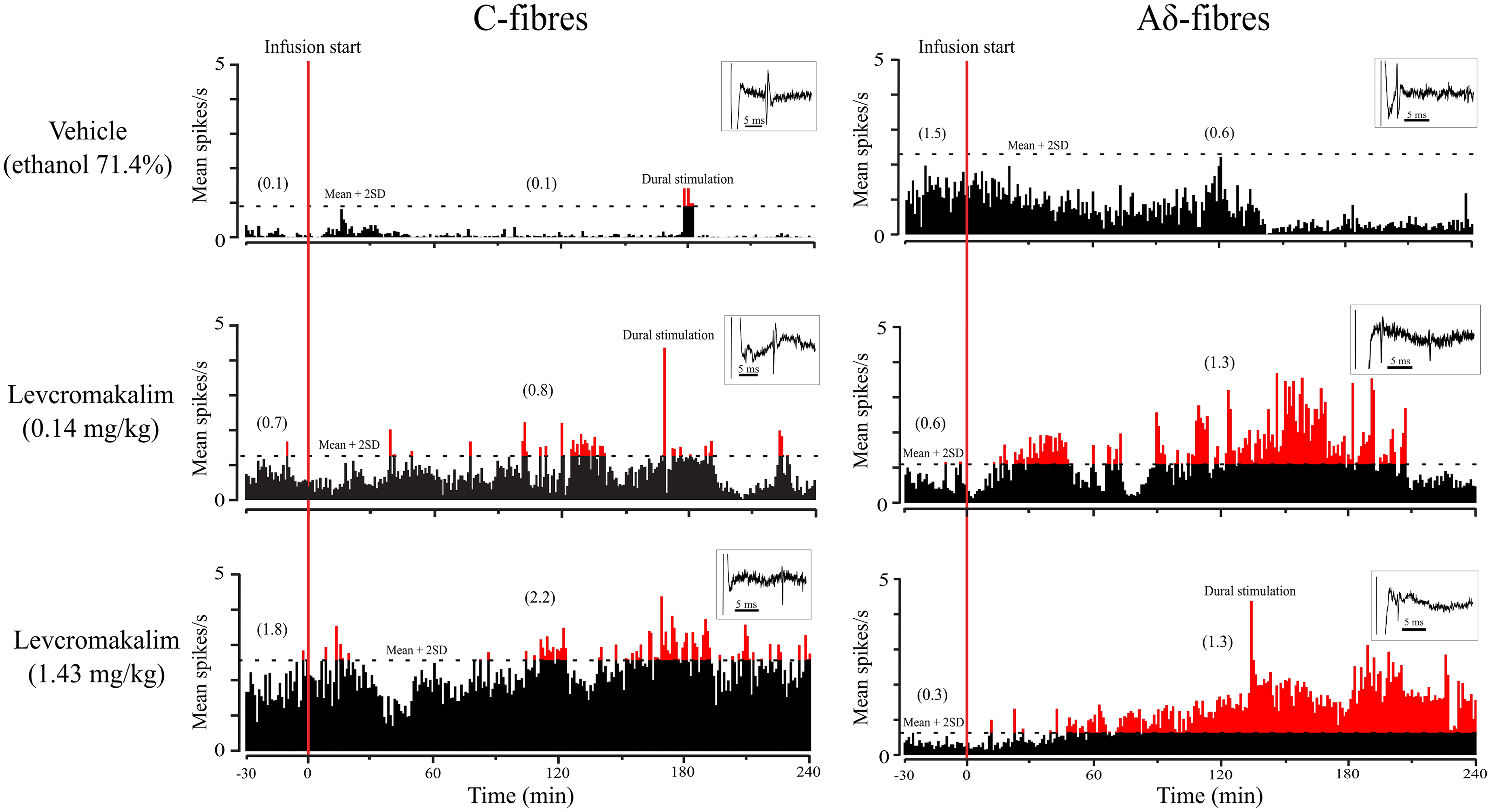
Firing rates of C- (left row) and Aδ type (right row) meningeal nociceptors before and after intracarotid infusion of vehicle (ethanol 71.4%) or levcromakalim at doses of 0.14 mg/kg or1.43 mg/kg (from top to bottom row). The horizontal dotted line marks the firing level two standard deviations above the mean baseline rate. Mean firing rates are shown before and after start of infusion. Inserts show the neuronal spikes in response to dural electrical stimulation for the respective neurons.
Baseline firing rates
The median baseline firing rate across all recorded neurons was 0.72 spikes/s (IQR = 0.44–1.51). Firing rates were similar between Aδ-fibers (0.83 [IQR = 0.54–1.76] spikes/s) and C-fibers (0.70 [IQR = 0.33–1.19] spikes/s; p = 0.41). Neurons exposed to 1.43 mg/ml/kg of levcromakalim had a median baseline firing rate of 0.72 (IQR = 0.55–1.66) spikes/s, which did not differ from the vehicle group (0.59 [IQR = 0.39–1.72] spikes/s; p = 0.32). At a lower dose (0.14 mg/ml/kg), firing rates remained comparable to vehicle (0.82 [IQR = 0.39–1.72] vs. 0.59 [IQR = 0.39–1.72] spikes/s; p = 0.73). Baseline activity also did not differ significantly between neurons later classified as activated and those that remained inactive (0.61 [IQR = 0.37–0.96] vs. 0.93 [IQR = 0.50–1.70] spikes/s; p = 0.39).
Activation of meningeal nociceptors by levcromakalim
Levcromakalim at 1.43 mg/kg activated 9 (69%) of 13 meningeal nociceptors, compared with one (11%) of nine in the vehicle group (p = 0.01) (Figure 3; see also supplementary material, Figures S1 and S2). Activation rates did not differ significantly between Aδ fibers (6 of 8; 75%) and C fibers (3 of 5; 60%; p = 1.00) (Table 1; Figure 4; see also supplementary material, Figure S3). Likewise, activation was comparable in males (4 of 7, 57%) and females (5 of 6; 83%; p = 0.61). Due to the lack of sex-related differences, data from both sexes were pooled to improve statistical power and generalizability. The mean activation duration following levcromakalim was 93.7 ± 47.7 minutes. In five of nine activated neurons, firing persisted until the end of the recording.

Fold increase in firing rate relative to baseline in neurons activated by levcromakalim (1.43 mg/kg) compared to vehicle (ethanol 71.4%). ***p < 0.001. **p < 0.01. *p < 0.05.

Firing rate in fold increase relative to baseline amongst neurons activated by levcromakalim (1.43 mg/kg), separated according to fiber type.
Summary of results for levcromakalim (1.43 mg/kg).

For activated neurons.
Number of bins above baseline mean ± 2 SD.
IQR = interquartile range; NA = not applicable.
At a lower dose (0.14 mg/kg), levcromakalim activated three of 14 neurons (21.4%), all Aδ fibers. This rate did not differ significantly from vehicle (p = 0.56) or between fiber types (p = 0.20) (Table 2).
Summary of results for levcromakalim (0.14 mg/kg).

For activated neurons.
Number of bins above baseline mean ± 2 SDs.
IQR = interquartile range; NA = not applicable.
Neuronal response latency and firing patterns after levcromakalim at 1.43 mg/kg
For response latency, activated neurons exhibited a median latency of 103 minutes (IQR = 53–144). Latency did not differ by fiber type: Aδ fibers activated at 113 minutes (IQR = 42–114) and C fibers at 92 minutes (IQR = 82–111; p = 1.00).
Spontaneous firing rates increased significantly across all activated neurons, rising by 60% from a baseline of 0.67 spikes/s (IQR = 0.55–0.96) to 1.12 spikes/s (IQR = 0.70–2.01; p = 0.040). Firing rates in activated neurons were significantly increased at one hour post-infusion (p = 0.003) and remained elevated at two (p < 0.001), three (p = 0.002) and four hours (p = 0.046) (Figure 3). When all recorded neurons were analyzed, regardless of activation status, firing rates increased significantly at two (p = 0.037) and three hours (p = 0.046). No significant changes were observed at one (p = 0.052) or four hours (p = 0.255) (see supplementary material, Figure S3).
The median number of spikes exceeding the activation threshold across all responsive neurons was 2301 (IQR = 1251–5371). There was no significant difference between activated Aδ fibers (median = 4997; IQR = 2708–9049) and C fibers (median = 1136; IQR = 553–1593; p = 0.10). Likewise, the number of spikes did not differ significantly between neurons in male animals (median, 4997; IQR = 3522–8250) and those in females (median, 2070; IQR = 1251–2300; p = 0.56).
Physiological effects of levcromakalim
Levcromakalim 1.43 mg/kg significantly increased heart rate by 9.0% at 10 minutes and by 6.8% at 20 minutes after start of infusion, compared with vehicle (both p < 0.01) (Figure 5; see also supplementary material, Table S1). Levcromakalim 0.14 mg/kg did not increase heart rate significantly compared with vehicle at any time point (Figure 5; see also supplementary material, Table S2).

Mean change in heart rate from baseline after levcromakalim (1.43 mg/kg and 0.14 mg/kg) and vehicle (ethanol 71.4%). Significantly greater increase in heart rate at 10 and 20 minutes after start of the 20-minute intracarotid infusion with levcromakalim (1.43 mg/kg) compared to vehicle. *p ≤ 0.05 compared to vehicle.
Discussion
Our single-unit electrophysiological recordings revealed that levcromakalim activates meningeal nociceptors within the trigeminal ganglion. Relying on a study design that closely mimicked human provocation studies, levcromakalim activated 70% of neurons at a latency of about 100 minutes, compared with 10% in the vehicle group. Activation rates were similar between Aδ- and C-fibers and did not differ by sex. These data bolster the concept that peripheral KATP channels contribute to the pathogenesis of headache and migraine.
Clinical correlates
Our findings help explain clinical observations showing that levcromakalim induces headache in healthy adults, migraine attacks in people with migraine, and cluster attacks in those with cluster headache.7,11–13 The delayed onset of migraine in patients, with a median of three hours, corresponds well to the 100-minute latency we observed for nociceptor activation. Likewise, the prolonged duration of headache, often lasting hours to days, aligns with sustained nociceptor firing, which frequently persisted until the end of the recording period. 7 Taken together, our observations suggest that activation of meningeal nociceptors provide a unifying mechanism by which KATP channel opening triggers headache across a spectrum of primary and secondary headache disorders.7,12,13
Possible mechanisms of action
The current results document that KATP channel opening activates meningeal nociceptors, likely explaining its headache-inducing properties in humans. A primary peripheral effect presumably triggers the activation of second- or third-order central trigeminovascular neurons, leading to the headache phase of migraine. The precise mode by which levcromakalim activates meningeal nociceptors remains unknown. However, two potential mechanisms warrant consideration:
Vessel-to-Neuron signaling (indirect effects via meningeal vasculature)
Levcromakalim activates KATP channel on VSMCs, causing potassium efflux and arterial relaxation.30,31 It induces sustained dilation of the middle meningeal artery in both rats and humans.11,32 These channels are also key effectors of meningeal vasodilation resulting from migraine-inducing compounds such as CGRP and PACAP-38.32,33 Both peptides activate cAMP signaling pathways that culminate in the opening of ATP-sensitive potassium channels, ultimately promoting relaxation of the VSMCs and meningeal arterial dilation. 32 Some findings suggest that, at least in coronary arteries, CGRP might exert vasodilatory effects through cAMP-independent mechanisms, 34 and it is likely that other signals related to cGMP or other intracellular cascades eventually culminate in the opening of KATP channels. After opening of the channels, mechanical distension and the rising potassium gradient near meningeal arteries are posited to activate perivascular meningeal nociceptors. 8 Although direct evidence for this cascade is lacking, it aligns with the observation that well-established migraine-inducing agents cause meningeal arterial dilation.11,35–37 That opening of KATP channels activates meningeal nociceptors might therefore support a common cascade for agents that trigger migraine. Likewise, vascular KATP channel contribute to the nocifensive response of mice exposed to levcromakalim. 15 Other lines of evidence also support such a cascade, including findings that levcromakalim triggers migraine attacks despite CGRP receptor blockade, suggesting a downstream site of action. 10 On the other hand, some animal behavioral studies have suggested that CGRP signaling is required for behavioral responses to levcromakalim.16,38,39 Nevertheless, in many of these studies it took several days of repeated exposure for levcromakalim to induce behavioral responses,16,38,39 raising concerns about their sensitivity and specificity. To reconcile such discrepancies, an important step forward would be to investigate whether CGRP receptor blockade prevents activation of meningeal nociceptors after levcromakalim.
The delayed effect of levcromakalim aligns with the delayed development of migraine in patients, as well as a similar delayed behavioral sensitivity to von Frey filaments in certain rodent studies.16,38 Nevertheless, the mechanisms underlying this temporal gap remains largely unresolved. One potential explanation might lie in the prolonged, hour-long cephalic vasodilation induced by levcromakalim. Rather than instantaneous activation, sustained vasodilation and potassium efflux might gradually promote the activation and recruitment of meningeal nociceptors. Eventually, the sum trigeminal input would then culminate in a migraine attack. Some lines of evidence support a role of such prolonged vasodilation, at least among other migraine provoking substances. For example, vasoactive intestinal peptide (VIP) induces only a short-lived cephalic vasodilation that resolves quickly once infusion is stopped. A short, 20-minutes infusion of VIP did not induce migraine, 40 but when the vasodilation was sustained with a two-hour infusion, VIP induced migraine in 71% of patients. 41 Moreover, interrupting this vasodilation with vasoconstrictors such as sumatriptan appears capable of preventing the development of migraine attacks after other migraine-inducing agents, such as PACAP-38. 42
Apart from a role of prolonged vasodilation, other potential mechanisms of action include as of yet unelucidated intracellular signaling cascades, 43 altered gene expression, 39 or secondary local responses within the meningeal neurovascular environment (e.g. related to neurogenic inflammation). 38 The mechanisms of such delayed responses to opening of KATP channels nevertheless remain to be more specifically deciphered.
Direct neuronal effects
Opening of neuronal KATP channels hyperpolarizes the membrane,7,17–19 which makes a direct neuronal mechanism seem unlikely as the cause of headache. By contrast to its headache-inducing effect, this hyperpolarization typically imparts antinociceptive properties in the central nervous system and even peripherally.20–26 These observations led to proposals that levcromakalim and similar potassium channel openers might treat acute and chronic non-cephalic pain and certain facial pain type.44,45 The discrepancy likely reflects fundamental differences between headache pathophysiology and other forms of pain. Some studies suggest that levcromakalim's hyperpolarizing effect might provoke headache through other processes.43,46 These include possible hyperpolarization of interneurons that inhibit trigeminal signaling, or prolonged hyperpolarization that eventually activates hyperpolarization-activated cyclic nucleotide-gated channels.43,46 Although we did not record membrane potentials, we did not observe inhibition of firing rate prior to the neuronal activation. These findings contradict the hypothesis that hyperpolarization alone explains levcromakalim-induced headache. Additional evidence also casts doubt on this idea. Some studies suggest KATP channel subunits are only minimally expressed within the trigeminal ganglion of humans and rodents, 15 and a neuronally acting KATP channel opener (NN414) failed to induce migraine attacks in people with migraine. 27 Moreover, opening other types of neuronal potassium channels (e.g. KCNQ2/3), which do not reside on VSMCs, blocks the activation of meningeal nociceptors. 47
Notably, although our results did not suggest differences in activation according to fiber type or sex, the sample sizes for these comparisons were limited. Future studies might therefore investigate whether there exist less marked differences in response properties. For example, across multiple measures C-fibers appeared to show a descriptively but insignificantly smaller response than Aδ-fibers.
Possible central effects
Levcromakalim is lipophilic and has a molecular weight of 286.3 Da, and so it is likely to cross the blood–brain barrier (BBB). 48 Magnetic resonance angiography data has shown that it dilates the middle cerebral artery in humans. 11 Although the extent of BBB penetration remains unclear, central effects could theoretically contribute to headache. Even though our data cannot confirm or rule out central sites of action, previous preclinical studies however suggest that levcromakalim exerts antinociceptive effects within the central nervous system.20–26 Coupled with the current findings, this suggests that peripheral actions are more likely to explain how levcromakalim induces headache and migraine. However, a primary peripheral effect likely triggers second- or third-order central trigeminal neurons, contributing to the clinical presentation of migraine attacks.
KATP channel subtypes and therapeutic potential
An outstanding question is whether specific KATP channel subtypes are more relevant to the headache-inducing properties of levcromakalim. KATP channels exist in two main subtypes: Kir6.1/SUR2B and Kir6.2/SUR2A. 49 Levcromakalim most strongly activates Kir6.1/SUR2B, 50 which is predominantly found on VSMCs in the rat meningeal artery and on neurons in the trigeminal ganglion. 30 Partly selective inhibitors of Kir6.1/SUR2B attenuate levcromakalim-induced dilation of the rat middle meningeal artery, 30 suggesting that Kir6.1/SUR2B is a prime target for treating migraine and other headache disorders. Human data shows that the non-specific KATP channel blocker glibenclamide does not reduce headache incidence or intensity or vasodilation following levcromakalim administration, suggesting that more specific Kir6.1/SUR2B blockade might be necessary. 51 Although glibenclamide prevented both behavioral responses and CGRP-induced vasodilation in animal,14,52 this is likely due to the substantially higher dosage of glibenclamide which was used in animals. Such dosages are unsuitable for human investigation due to glibenclamide's strong hypoglycemic effects, comprising a key caveat in animal behavioral studies, because hypoglycemic obtundation can non-specifically suppress responses such as hindpaw withdrawal.
There remains a need for agents that selectively block Kir6.1/SUR2B KATP channels. No such pharmacological drugs currently exist, although they represent promising candidates for drug development. By acting as downstream effectors of multiple signaling pathways, these channels may offer greater efficacy than treatments that target individual neuropeptides. Notably, selective KATP channel blockers might not require BBB penetration, which would reduce concerns related to central side effects.
Conclusions
Our results demonstrate that the opening of vascular KATP channels via intracarotid levcromakalim infusion activates meningeal nociceptors in a dose-dependent manner. This finding suggests that levcromakalim induces headache in healthy adults and migraine attacks in people with migraine by activating meningeal nociceptors. This effect is most likely indirect, making it prudent to prioritize the development and testing of vascular KATP channel blockers of the Kir6.1/SUR2B subtype as potential treatments for migraine and other headache disorders.
Clinical implications
Opening of ATP-sensitive potassium channels activated meningeal nociceptors implicated in migraine and other headache disorders
Similar activation rates were observed for Aδ and C-fibers, as well as between male and female animals.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251359237 - Supplemental material for Opening of ATP-sensitive potassium channels activates meningeal nociceptors: Implications for the origin of migraine headache
Supplemental material, sj-docx-1-cep-10.1177_03331024251359237 for Opening of ATP-sensitive potassium channels activates meningeal nociceptors: Implications for the origin of migraine headache by Rune H. Christensen, Andrew M. Strassman, Messoud Ashina, Håkan Ashina and Rami Burstein in Cephalalgia
Footnotes
Acknowledgements
We extend our sincere gratitude to Dr Agustin Melo-Carrillo for his dedicated teaching and supervision of surgery, anesthesia, ventilation and electrophysiological techniques during the present study. In addition, we thank Dr Melo-Carrillo for his important input on project execution and administration, and for having developed and graciously taught the innovative intracarotid approach, which was used for drug administration.
Data availability
Data supporting the presented findings are available upon reasonable request from a qualified investigator.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RHC has received personal fees from Pfizer and Teva, outside of the submitted work. AS declares no competing interests. MA has received personal fees from AbbVie, Amgen, AstraZeneca, Eli Lilly, GlaxoSmithKline, Lundbeck, Novartis, Pfizer and Teva, outside of the submitted work. MA is also an Associate Editor of Brain and The Journal of Headache and Pain. HA has received personal fees from AbbVie, Lundbeck, Pfizer and Teva, outside of the submitted work. HA is also an Editorial Board Member of The Journal of Headache and Pain. RB has received research grants from NINDS, AbbVie, Eli Lilly, Modulight and Teva. He also received personal fees from AbbVie, Biohaven, Escient, Neurolief, Percept, Progastro Kft, Rapport Therapeutics and Salvia Bioelectronics.
Ethical statement
All experiments were approved by the animal care committees of Beth Israel Deaconess Medical Center and Harvard Medical School. The study complied with the guidelines of the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by a research grant from the Lundbeck Foundation (R403-2022-1352 to HA) and National Institute of Neurological Disorders and Stroke (NINDS) grants NS128045 (RB).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.