
Abstract
Introduction
Effectiveness of candesartan in migraine prevention is supported by two randomized controlled trials. We aimed to assess the effectiveness, tolerability, and response predictors of candesartan in the preventive treatment of migraine.
Methods
Observational, multicenter, prospective cohort study. The 50%, 75% and 30% responder rates, between weeks 8–12 and 20–24, were compared with the baseline. Treatment emergent adverse effects were systematically evaluated. Response predictors were estimated by multivariate regression models.
Results
Eighty-six patients were included, 79.1% females, aged 39.5 (inter-quartile range [IQR] 26.3–50.3), with chronic migraine (43.0%), medication overuse headache (55.8%) and a median of two (inter-quartile range: 0.75–3) prior preventive treatments. At baseline patients had 14 (10–24) headache and 8 (5–11) migraine days per month. The 30%, 50% and 75% responder rates were 40%, 34.9% and 15.1% between weeks 8–12, and 48.8%, 36%, and 18.6% between weeks 20–24. Adverse effects were reported by 30 (34.9%) and 13 (15.1%) patients between weeks 0–12 and 12–24, leading to discontinuation in 15 (17.4%) patients. Chronic migraine, depression, headache days per month, medication overuse headache, and daily headache at baseline predicted the response between weeks 20–24.
Conclusion
Candesartan effectiveness and tolerability in migraine prevention was in line with the clinical trials’ efficacy.
Introduction
Candesartan is a benzimidazole-derived angiotensin II receptor antagonist. It competes with angiotensin II for the binding of the angiotensin II receptor subtype 1, blocking angiotensin II-mediated vasoconstriction and inducing vasodilation (1). It was developed in the early nineties, initially named TCV-116 (2), and then CV-11974 (3). In July 2010, the European Medicines Agency recommended candesartan for essential hypertension or heart failure and impaired left ventricular systolic function (4). Since then, it has also been extensively used in the treatment of diabetic nephropathy and coronary artery disease (4–7), and more recently, migraine prevention.
Efficacy of candesartan in migraine prevention was determined by two randomized controlled trials (RCTs) (8,9). The first study, conducted between 2001 and 2002, included patients with two to six migraine attacks per month and prior use of 0–1 preventive drugs. Sixty patients were randomized to placebo or candesartan (16 mg), and 46 patients completed the study. In the intention-to-treat (ITT) analysis, patients treated with candesartan showed 4.9 (standard deviation (SD) 10.6) fewer headache days during the 12-week treatment period (8). The second study was a double-blind cross-over trial with three periods, including placebo, candesartan (16 mg) or propranolol (160 mg). The study was completed between 2009 and 2012 and included patients with 2–14 headache days per month and prior use of 0–2 preventive drugs. Fifty-four patients completed the study. During the 12-week treatment period, patients treated with candesartan showed a reduction in monthly headache days of 2.91 (SD 1.06) days (9).
Evidence for the use of candesartan for chronic migraine (CM) or treatment-resistant migraine patients came from two observational studies (10,11). The first one retrospectively evaluated patients treated between 2015 and 2018. Out of 253 patients, 81 were included in the final analysis: 84% reported CM at baseline and used a median of three prior preventive drugs (10). The study showed that 44% of patients showed some degree of response, defined as a reduction in the number of headache days, migraine days, or the severity of migraine attacks (10). Another retrospective cohort study was conducted between 2008 and 2019 (11). This study included 120 patients, 70% of them with CM at baseline, with a mean number of 3.7 prior preventive drugs. Patients presented with 4.3 fewer headache days per month between weeks 8–12, compared with baseline (11). In addition, these studies showed preliminary data about some possible response predictors, including younger age (10), and a longer disease duration was associated with a higher likelihood of response (10), while those with daily headache (10,11) and a higher number of prior preventive drugs (11) were less likely to respond.
In the RCTs, the tolerability profile was emphasized as one of the main strengths of candesartan. Three patients discontinued candesartan due to dizziness, compared to one patient in the placebo group, with no statistically significant between-group differences (8). In the second RCT, patients treated with candesartan showed a higher frequency of dizziness (50% of patients), with no statistically significant differences in the rest of the reported adverse events (AEs) (9). In the second observational study, 18% of patients discontinued candesartan due to inadequate tolerability, with dizziness and hypotension as the most frequently reported AEs (11).
After these four studies, several questions remained unanswered, including the mid-term effectiveness and tolerability, the effectiveness in patients with CM or difficult-to-treat patients, and whether some patients had a higher/lower probability of response. The main objective of this study was to evaluate the percentage of migraine patients who exhibit at least a 50% response rate to candesartan in headache day reduction, evaluated in the period between weeks 20 to 24, compared with baseline.
Methods
Study design
CandeSpartan was a prospective, multicenter cohort study. The study protocol was published at ClinicalTrials.gov (NCT04138316). The study was reported in accordance with the Strengthening The Reporting of Observational Studies in Epidemiology (STROBE) checklist (12). The study was approved by the Ethics Review Board (PI 19-1452). All patients provided written informed consent.
Setting
The study was conducted at six third-level reference headache centers in Spain, specifically in Barcelona (n = 1), Madrid (n = 2), Palma de Mallorca (n = 1), Santander (n = 1), and Valladolid (n = 1). The study period was between 2 January 2020 (first patient, first visit) and 16 December 2022 (last patient, last visit). Follow-up lasted six months, and patients had three in-person visits: baseline, three months, and six months. Data were collected through standardized questionnaires.
Participants
Patients were included if they: 1) had a diagnosis of migraine according to the International Classification of Headache Disorders (ICHD), 3rd version, criteria (13); 2) were treated with candesartan under the prescription criteria of the physician and according to local guidelines (14); 3) were aged 18 years or older; 4) had four or more headache days per month, in the preceding three months; 5) fulfilled migraine ICHD-3 criteria for at least 12 months prior to baseline; 6) had a migraine onset before the age of 50; 7) were able to provide informed consent and agreed to participate.
The exclusion criteria were: 1) previous failure of three or more preventive drugs, defined as insufficient effectiveness after being used at a sufficient dose and for an adequate duration, or inadequate tolerability; 2) concomitant use of another drug with a possible migraine-preventive profile, or its recent use (less than five half-lives); 3) history of another primary headache disorder with a frequency of ten or more headache days per month at baseline; 4) continuous daily headache in the month prior to inclusion in the study; 5) pregnancy or breastfeeding; 6) any relevant cardiovascular disorders; 7) kidney diseases; 8) hyperkalemia; 9) prior use of candesartan; 10) current use of another angiotensin-conversing enzyme inhibitor or angiotensin II receptor antagonist; 11) alcohol or illicit drug misuse.
Screening and sampling
The recruitment method was non-probabilistic, based on an opportunistic recruitment.
Study objectives
The study endpoints were pre-specified and published at ClinicalTrials.gov on 24 October 2019, prior to the study onset. The objectives were selected based on the Guidelines of the International Headache Society for controlled trials of preventive treatment of CM in adults. The primary endpoint was to evaluate the 50% responder rate between weeks 20–24, with respect to the baseline period.
The secondary endpoints included: 1) to determine which demographic or clinical variables present at baseline are associated with a 50% reduction in the frequency of headache days per month between weeks 20 to 24 with respect to the baseline period; 2) to assess which variables present at baseline are associated with a higher reduction in the number of headache days per month (HDM) between weeks 20 to 24, with respect to the baseline period; 3) to evaluate the percentage of patients who present with a 30%, 50%, 75% or 100% response rate to candesartan in headache day reduction evaluated in the periods between weeks 8 to 12 and 20 to 24 compared with baseline; 4) to assess the change in HDM, migraine days per month (MDM), acute medication days per month (ADM) and triptan use days per month (TDM) between weeks 8 to 12 and 20 to 24 compared with baseline; 5) to determine frequency and type of AEs, and the percentage of patients who discontinue candesartan due to treatment-emergent adverse effects (TEAE).
The rationale for the evaluation between weeks 8–12 and 20–24, and not during the entire 12- or 24-week period was that with this being an observational study, the candesartan dose tapering protocols varied across the study sites, potentially resulting in different amounts of time to reach the target and stable dose. In addition, there was no prior information about whether candesartan had a fast onset.
Study intervention and procedures
Candesartan was prescribed as per the local standard of care (14). All study data were retrieved through a clinical interview with a headache expert and the review of medical records through a pre-defined data collection form and study questionnaires. Vital signs assessment and physical examination were performed by a neurologist. Headache diaries were given to the study participants. Patients were trained on how to complete the diaries, and they were instructed to complete the baseline headache diary for 30 days prior to candesartan use.
Variables
Demographic, clinical and safety data were collected. Demographic variables included sex, age, age of onset of migraine, weight, height, presence of overweight (body mass index (BMI) >25). Clinical variables collected at the baseline visit encompassed the type of migraine (episodic (EM) versus CM), the presence of aura, bilateral or unilateral predominance of headache, number of prior preventive drugs, treatment-resistant migraine criteria (15), medication overuse headache (MOH) according to the ICHD-3 criteria, headache intensity (0–100 verbal analog scale), and prior history of psychiatric comorbidity (anxiety, depression).
At all study visits, including the baseline visit, the headache profile was assessed according to pre-filled headache diaries and was measured by HDM, MDM, ADM and TDM. Blood pressure (mmHg) was measured in the right arm after five minutes of rest, and heart rate (beats per minute) was measured. In addition, the presence of allodynia was assessed, according to the Allodynia Scoring Checklist – 12 (ASC-12) scale (16). This instrument includes twelve questions assessing daily activities and evaluates the presence of pain or unpleasantness on a 0–2 scale (never/not applicable, less than half of the time, half the time or more), with a total score between 0 and 24 where three or more points indicates allodynia, five points indicates moderate allodynia, and nine or more points indicates severe cutaneous allodynia (16). The adverse headache impact was assessed by the Headache Impact Test – 6 (HIT-6), which includes six questions that are scored between 6 and 13, with a final score between 36 and 78. The score is stratified into four categories: little or no impact (<50), some impact (50–55), substantial impact (56–59) and severe impact (>59) (17); thus, a higher score indicates a higher disability attributed to headache. The presence of clinically relevant anxiety and/or depression was assessed with the Hospital Anxiety and Depression Scale (HADS), which includes twelve questions assessed on a 4-point Likert scale (0–3), where a total score between 8–10 indicates moderate anxiety/depression, and a score >10 suggests severe anxiety/depression. Safety variables included dizziness, tiredness, reduced physical capacity, nausea, constipation, sleep disturbance, sexual dysfunction, and paresthesia.
Data sources and measurements
Structured and standardized data collection instruments were used to ensure the consistency of assessments across the different study sites. Paper headache diaries were given to the study participants to collect the clinical variables throughout the entire study period, and patients were trained in their correct use prior to the treatment onset. In the evaluation of AEs, patients were first asked to spontaneously report any possible AE. Afterwards, a structured questionnaire was administered, and patients were asked to describe whether they experienced any of the listed AEs. The AEs that were systematically evaluated were based on a prior RCT (9) and included dizziness, tiredness, reduced physical capacity, nausea, constipation, sleep disturbances, sexual dysfunction, paresthesia, and others. The severity of the AE and the relationship to the study drug were determined by the investigators. The study database was created in RedCap.
Bias
A series of biases were anticipated in the study design and completion. Details are available in the Online Supplementary Material.
Study size
According to the results of both previous RCTs (8,9), in the ITT groups, 18/57 and 24/66 patients had a 50% responder rate, for a combined rate of 42/123 = 34.1%. In the placebo group the 50% response rate was 1/57 and 14/64, for a combined rate of 15/121 = 12.4%. For an alpha value of 5%, and statistical power of 90%, with an anticipated loss of 10% patients, the required sample size for this study was estimated to be 86 patients.
Statistical methods and variables presentation
Qualitative and ordinal variables are presented as frequencies and percentages. Continuous variables are represented as means and SD or medians and inter-quartile range (IQR) depending on the type of distribution. The normality of the sample distribution was assessed with the Kolmogorov-Smirnov test. Both ITT and per-protocol (PP) analyses were conducted.
The 30%, 50%, 75% and 100% responder rates were calculated at weeks 8–12 and 20–24, compared to baseline (18). To evaluate the change in the HDM, MDM, ADM and TDM at weeks 8–12 and 20–24, with respect to baseline, paired t-tests or Kruskal Wallis tests were used, depending on the type of distribution. Pearson or Spearman’s coefficients were calculated for continuous variables.
To evaluate the predictors of responsiveness, two statistical models were developed. First, a logistic regression model was created, with the 50% responder rate between weeks 20–24 as the dependent variable. All variables with a P value <0.2 in the univariate regression analysis were included in a multivariate regression analysis, using backward Wald stepwise elimination of variables (19). These results are presented as odds ratios (OR) and 95% confidence intervals (CI). Second, a linear regression model was performed to assess the change in the number of HDM from baseline to weeks 20–24; an ITT method and baseline observation carried forward (BCF) were used. First, a univariate regression analysis was done, and variables that presented a P value <0.2 in the univariate regression were included in a multivariate regression analysis (19). The raw values are presented, and then a backward procedure was employed to select the variables that better predicted the change in HDM. These results are represented as the β regression coefficient with 95% CI.
Multiple comparisons were handled with a false-discovery rate (FDR), according to the Benjamini-Hochberg procedure. To minimize the overestimation of the responses and the risk of false positive results, missing data were managed in the most conservative way, by both BCF and last observation carried forward (LOCF). The variables were visually inspected, and imputation was done manually taking into account the baseline parameter or the last observation. Statistical analysis was done with SPSS v.26 (IBM Corp, Armonk, USA).
Results
Recruitment was stopped once the target sample size of 86 patients was achieved. Details about the number of patients recruited per site is available in the Online Supplementary Material. Table 1 summarizes the demographic and clinical variables of the study sample at baseline. The most frequently employed prior preventive drugs were topiramate (n = 41, 47.7%), amitriptyline (n = 40, 46.5%), beta blockers (n = 28, 32.6%), and flunarizine (n = 17, 19.8%).
Demographic and clinical variables at baseline.

Candesartan dose
Between weeks 0–12, patients used either 4 mg (n = 12, 13.9%), 8 mg (n = 47, 54.7%), 12 mg (n = 7, 8.1%), 16 mg (n = 19, 22.1%), or 36 mg (n = 1, 1.2%) of candesartan. By weeks 12–24, 20 (23.3%) patients had discontinued candesartan. The reasons for treatment discontinuation were inadequate tolerability (n = 15, 17.4%), follow-up discontinuation (n = 2, 2.3%), study consent withdrawal (n = 1, 1.2%), change of country of residence (n = 1, 1.2%), and change of city of residence (n = 1, 1.2%). Figure 1 shows the survival curve of the persistence of candesartan use. The remaining 66 patients used either 4 mg (n = 1, 1.2%), 8 mg (n = 30, 34.9%), 12 mg (n = 11, 12.8%), 16 mg (n = 22, 25.6%), 20 mg (n = 1, 1.2%) or 24 mg (n = 1, 1.2%). Online Supplementary Figure 1 displays the candesartan maximal dose received per patient.
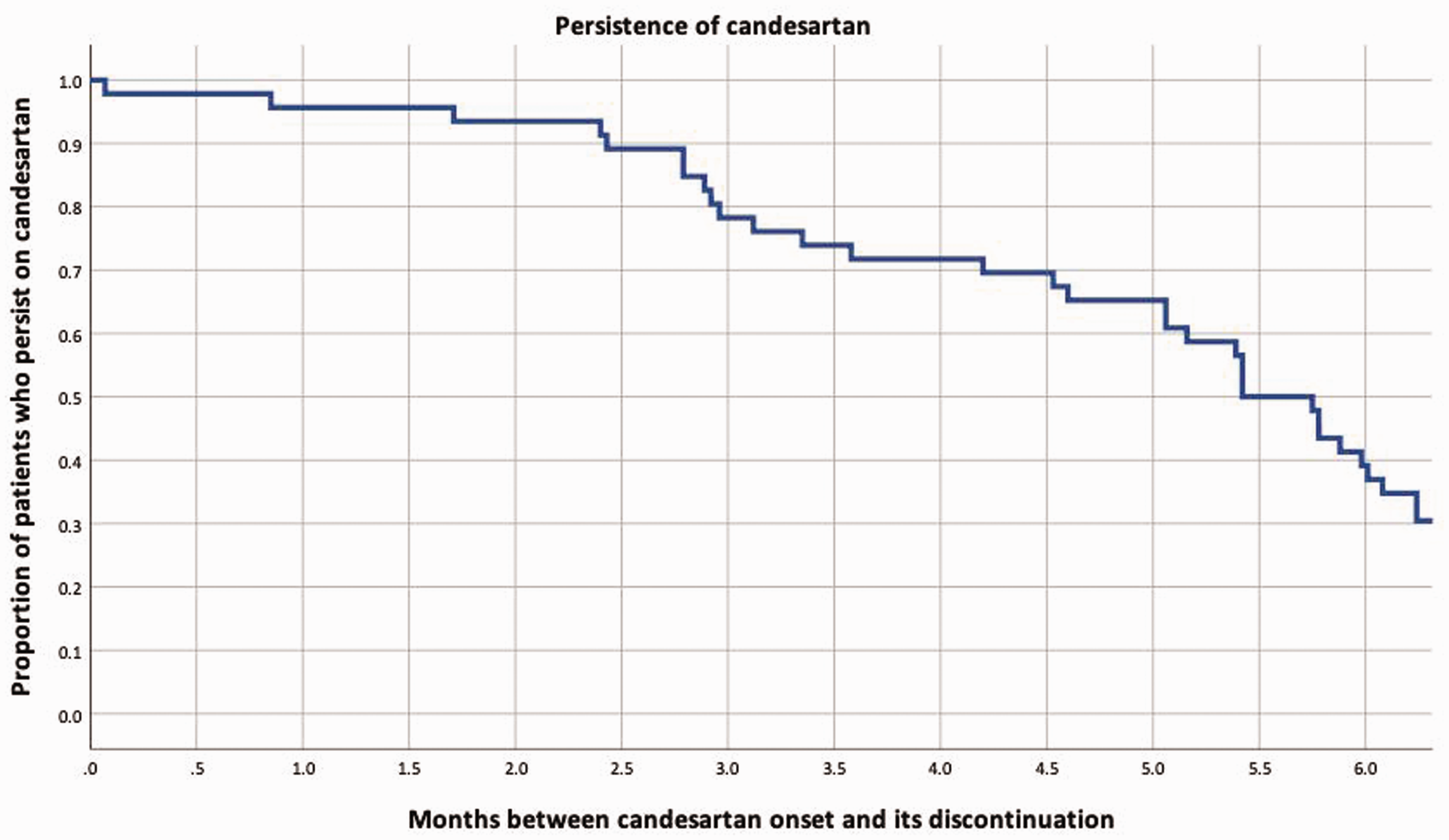
Persistence of candesartan use during the entire study period.
Change in headache/migraine/acute medication/triptan days per month
There was a statistically significant reduction in the number of HDM, MDM, ADM and TDM from baseline to weeks 8–12 and a reduction in HDM, MDM and ADM from baseline to weeks 20–24 (Online supplementary Figure 1). Online Supplementary Table 1 and Supplementary Figures 2 and 3 show the relationships between the number of HDM at baseline versus weeks 8–12 and HDM at baseline versus weeks 20–24.
Primary endpoint: 50% response rate at weeks 20–24
The proportion of patients who achieved a 50% response rate in HDM was between 36.0% and 47.0%, depending on the type of analysis (ITT vs. PP, respectively).
Responder rates
Figure 2 shows the 30%, 50%, 75% and 100% responder rates.
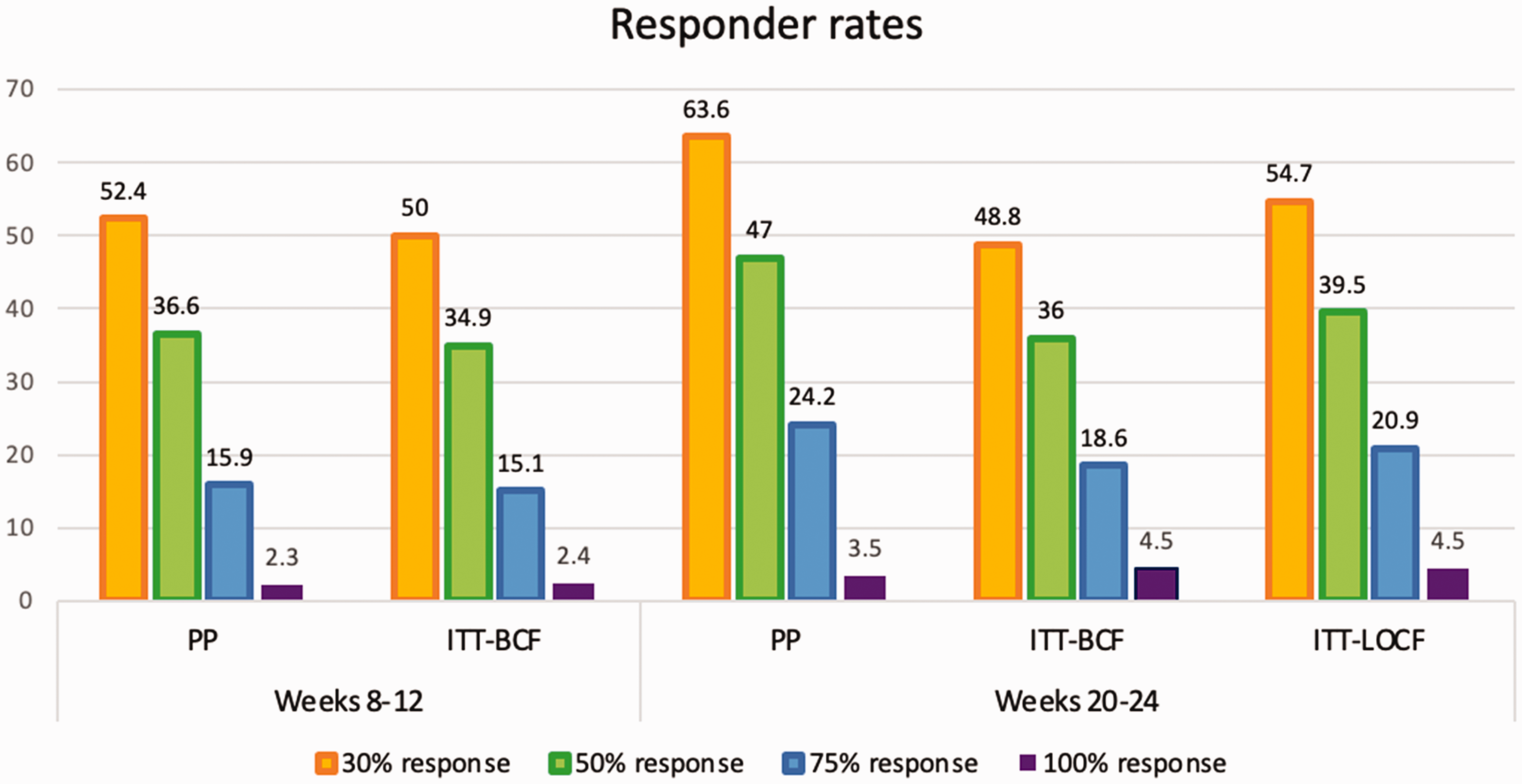
30% (yellow), 50% (green), 75% (blue) and 100% (purple) responder rates. PP: Per protocol. ITT: intention-to-treat. BCF: Baseline observation carried forward. LOCF: Last observation carried forward. Y axis: Percentage of patients.
Tolerability
The number of patients who experienced at least one TEAE was 30 (34.9%) during weeks 0–12 and 13 (15.1%) during weeks 12–24. The most frequently reported AEs between weeks 0–12 and 12–24 were lightheadedness (19.8%, 8.1%) and asthenia (16.3% and 3.5%). Figure 3 shows the frequency, type, and severity of TEAE.
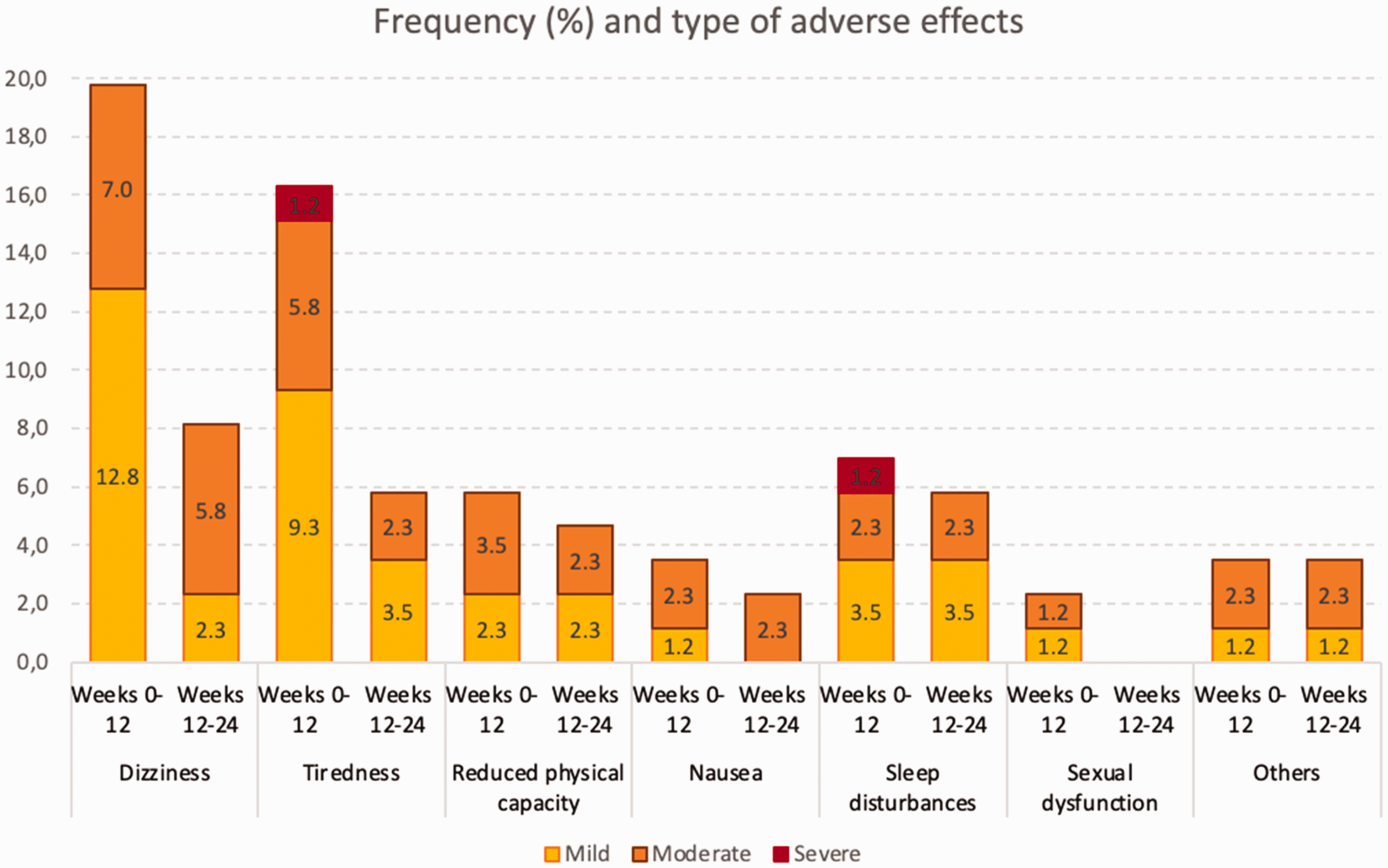
Frequency (percentage of patients over the total number of patients) and type of adverse effects. Y axis: Percentage of patients.
The number of patients who experienced an AE leading to treatment discontinuation was 15 (17.4%), including lightheadedness (n = 5, 5.8%), sleep disturbances (n = 5, 5.8%), asthenia (n = 3, 3.5%), nausea (n = 2, 2.3%), constipation (n = 1; 1.2%), and sexual dysfunction (n = 1; 1.2%)
Response predictors
In the univariate regression analysis (Online Supplementary Table 2), both a higher number of HDM and ADM at baseline as well as presence of CM at baseline were associated with a higher probability of achieving a 50% response rate at weeks 20–24.
In the multivariate logistic regression analysis, a higher number of HDM at baseline was associated with a higher probability of response (OR: 1.093; 95% CI: 1.023–1.168; p = 0.009), while a lower number of TDM was not associated (OR: 0.916; 95% CI: 0.827–1.014; p = 0.089).
In the ITT-BCF multivariate linear regression analysis, the variables that showed a statistically significant association with a higher change in the number of HDM from baseline to weeks 20–24 were HDM at baseline (standardized B: 0.530; 95% CI: 0.0386–0.830; p < 0.001) and the allodynia symptom score at baseline (standardized B: −0.262; 95% CI: −1.227–0.184; p = 0.009). Full details of the linear regression analysis are available in Online Supplementary Table 3.
Discussion
In the era of novel preventive drugs for treating migraine, we aimed to revisit the effectiveness and tolerability of an “older” preventive drug. This was deemed necessary, given that only two RCTs had assessed its efficacy, and these included mostly treatment-naïve EM patients (8,9). To this end, a multicenter prospective cohort study was designed and successfully completed. In the present study, half of the enrolled patients had MOH, almost half had CM, 74% were severely impacted by migraine, and 32% of patients fulfilled criteria for treatment-resistant migraine (15). In this “more difficult” to treat population, both effectiveness and tolerability were in line with prior studies (8–11).
Regarding effectiveness, assessed as the 50% response rate in HDM, this study’s results, where 35–47% of patients achieved a reduction of at least 50% in the number of HDM from baseline to weeks 8–12 or 20–24, were similar to those of the two RCTs – 31.6% and 43% (8,9), and to those of the retrospective cohort study - 32.5% (11). The lack of a placebo control group represents a significant limitation, since some patients could have responded by chance. In this regard, in the RCTs, the proportion of patients who achieved a 50% response compared with placebo was 1.8% and 23%, respectively. In our sample, the probability of a response due to random chance may be even lower, given the higher frequency of prior preventive drugs and the inclusion of patients with CM and daily headache, but this cannot be known with absolute certainty. This is the first study to calculate 30% and 100% responder rates, and the analysis used two different approaches, including only patients who maintained candesartan (PP), and the entire study sample (ITT). In the most conservative assessment (ITT analysis and BCF imputation method for the missing data), the proportion of patients who achieved at least a 30% responder rate was 50% at the third month (weeks 8–12) and 49% at the sixth month (weeks 20–24). This shows realistic and transparent results, which are not that different from those observed in other “old” oral preventive drugs, such as amitriptyline (20), flunarizine (21), and topiramate (22), where the absolute difference of patients who achieved a 50% response rate in monthly migraine days, compared to placebo, was 16.5% higher following amitriptyline and 16.8% higher following topiramate.
The proportion of patients who discontinued candesartan due to inadequate tolerability was 17.4%, which is higher than observed in the first RCT (10%) (8), and similar to what was reported in the retrospective cohort study (18.3%) (11). The proportion of patients who reported at least one TEAE was 35%, which is between that observed in the first RCT (46%) (8) and the second RCT (30%) (9). In addition, the tolerability profile was quite unique and not that different from that observed in the more novel preventive drugs (23). The most frequently reported AEs in this study were dizziness and tiredness, as expected, which are probably related to decreased blood pressure. This makes candesartan a drug to be considered in patients at risk of the consequences of central somnolence or cognitive symptoms. An unanswered question from our study is whether the probability of experiencing at least one AE was associated with the candesartan dose. In both RCTs the employed dose was 16 mg (8,9), while in our study most patients received a lower dose.
A higher number of HDM at baseline was associated with a higher likelihood of response, both in the logistic and linear regression analyses. In addition, a higher score in the allodynia scale at baseline was associated with a lower likelihood of response. Prior studies have suggested that daily headache (10,11), patients’ age (10), disease duration (10) and prior number of preventive drugs (11) are also associated with likelihood of response, which were not replicated in our study, perhaps due to the limited sample size.
One of the most interesting findings of the study is that candesartan seemed effective in patients who have been excluded in the most recent RCTs, which mostly evaluated patients treated with anti-calcitonin gene-related peptide drugs; patients in this study had CM (43%), MOH (56%), daily headache (10.5%), treatment resistant migraine (32%), clinically relevant anxiety (28%), clinically relevant depression (6%), allodynia (42%) or a severe impact of migraine disease (74%). Given the adequate tolerability profile, candesartan may be considered in these patients.
This study has relevant limitations that should be taken into consideration when interpreting its results. First, because of the absence of a control group, it cannot be ruled out that the observed effect is due to additional factors, other than the use of candesartan, such as regression towards the mean. Second, despite being trained, patients could have erroneously classified migraine episodes as no-migraine headache episodes, so the number of migraine attacks could potentially be under-estimated. Third, the study sample size could be underpowered to detect infrequent events or small differences, which may be relevant for the safety aspects and the response predictors. Fourth, patients with prior failure to more than three preventive therapies were not included in the study. Future studies should analyze whether candesartan is also effective in the treatment of more treatment-resistant patients too. Some other unanswered questions include how fast the effect of candesartan is perceived by patients, which may influence the results. In our study, the endpoints were assessed between weeks 8–12 and 20–24 and not during the entire treatment period, which could have influenced the results.
In conclusion, the present study provides class II evidence on the effectiveness of candesartan in the preventive treatment of migraine in a real-world setting. In this difficult-to-treat population, candesartan showed similar results to those observed in other pivotal clinical trials. The drug was well tolerated, and the observed AEs seemed drug-specific and mediated by the vascular effect of the drug. Candesartan should be considered as an additional weapon in the fight against migraine.
Article highlights
Candesartan was effective in the preventive treatment of episodic and chronic migraine in a real-world setting. One in six patients discontinued candesartan due to poor tolerability. The tolerability profile seems unique, with most adverse events related to its effect on blood pressure.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024241248833 - Supplemental material for CandeSpartan Study: Candesartan Spanish Response-prediction and Tolerability study in migraine
Supplemental material, sj-pdf-1-cep-10.1177_03331024241248833 for CandeSpartan Study: Candesartan Spanish Response-prediction and Tolerability study in migraine by David García-Azorín, Cristina Martínez-Badillo, Javier Camiña Muñiz, Ana Beatriz Gago-Veiga, Noemi Morollón Sánchez, Vicente González-Quintanilla, Jesús Porta-Etessam, Alvaro Sierra-Mencía, Nuria González-García, Yésica González-Osorio, Marcos Polanco-Fernandez, Andrea Recio-García, Robert Belvis Nieto and Angel Luis Guerrero-Peral in Cephalalgia
Footnotes
Acknowledgements
The authors gratefully acknowledge the study participants and their relatives.
Authors contributions
DGA, JCM, ABGV, NMS , VGQ, JPE and ALGP designed and conceptualized the study, CMB, JCM, ABGV, NMS, VGQ, JPE, ASM, YGO and ARG acquired the data, DGA analyzed the data, DGA and CMB drafted the manuscript; DGA, CMB, JCM, ABGV, NMS, VGQ, JPE, ASM, NGG, YGO , MPF, ARG, RBN and ALGP revised the manuscript for intellectual content. All authors read and approved the final manuscript.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DGA received honoraria for lectures/presentations from Abbvie/Allergan, Eli Lilly, Teva, Lundbeck, and Novartis. DGA Participated in clinical trials as the principal investigator for Pfizer, BioHaven and Lundbeck. DGA is junior editor of The Journal of Headache and Pain and Neurological Sciences. DGA received honoraria from the World Health Organization as subject matter expert.
ABGV received honoraria for lectures/presentations from Novartis, Lilly, Teva, Exeltis, Chiesi, Abbvie, Pfizer and Lundbeck.
ÁLGP received honoraria for lectures/presentations from Abbvie/Allergan, Eli Lilly, Teva, Lundbeck, and Novartis. ALGP Participated in clinical trials as the principal investigator for Eli Lilly, Teva, Abbvie, Novartis, Amgen and Lundeck.
The rest of the authors report no conflicts of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.