
Abstract
Objective
To investigate the efficacy and safety of injecting onabotulinum toxin A (BTA) towards the sphenopalatine ganglion (SPG) using the MultiGuide® in patients with persistent idiopathic facial pain (PIFP).
Methods
This cross-over, exploratory study compared the injection of 25 units BTA versus placebo in patients who met modified ICDH-3 criteria for PIFP. Daily pain diaries were registered for a 4-week baseline, a 12-week follow-up after each injection, and an 8-week conceptual washout period in between. The primary efficacy endpoint was the change from baseline to weeks 5–8 in average pain intensity using a numeric rating scale. Adverse events were recorded.
Results
Of 30 patients who were randomized to treatment, 29 were evaluable. In weeks 5–8, there was no statistically significant difference in average pain intensity between BTA versus placebo (0.00; 95% CI = −0.57 to 0.57) (P = 0.996). Following both BTA and placebo injections, five participants reported at least a 30% reduction in average pain during weeks 5–8 (P = 1.000). No serious adverse events were reported. Post-hoc analyses indicated a possible carry-over effect.
Conclusions
Injection of BTA toward the SPG with the MultiGuide® did not appear to provide a reduction in pain reduction at 5–8 weeks, although this finding may be influenced by a carry-over effect. The injection appears to otherwise be safe and well-tolerated in patients with PIFP.
Introduction
Persistent idiopathic facial pain (PIFP) is a persistent facial and/or oral pain condition (1,2). Pain may be described as either deep or superficial and is usually unilateral, but bilateral presentations have been described (3,4). PIFP is rare with an estimated prevalence of 4.4 per 100 000 persons (5,6). Symptoms are often diffuse, and many patients are both misdiagnosed and treated by several specialities without success or with harmful consequences (7). In one study (8), there was an average diagnostic delay of 31.6 months, seven consultations (median) were performed before a correct diagnosis was reached, and five ineffective prescriptions were issued. Patients with PIFP are prone to receive invasive dental treatment before final diagnosis (7–10). These patients are frequently referred to neurologists and maxillofacial surgeons and, as such, the condition is included in both the International Classification of Headache Disorders III (ICHD-3 III) (1) and, more recently, in the International Classification of Orofacial Pain, 1st edition (ICOP) (2).
It has been suggested that the sphenopalatine ganglion (SPG) and the parasympathetic system play a role in modulating the pain in PIFP and other pain syndromes in the area innervated by the trigeminal nerve (11–14). Parasympathetic nerves can be blocked by onabotulinum toxin A (BTA). Previous pilot studies on chronic migraine (15), cluster headache (16) and neuropathic facial pain (17) have indicated a potential effect on blocking the SPG with BTA. The aetiology of neuropathic facial pain and PIFP might be overlapping, and thus we wanted to investigate the effect of blocking SPG in patients suffering from PIFP. Our group has developed an instrument (MultiGuide®) to deliver medicines with high precision to the SPG with computed tomography-guided navigation. The aim of the present study was to investigate whether blocking the SPG with BTA using the MultiGuide® could give pain relief in patients with PIFP.
Methods
Study design
The study was a single-center, randomized, triple-blind, placebo-controlled cross-over, exploratory trial (18). It was a collaboration project between the Department of Neuromedicine and Movement Science (INB), Faculty of Medicine, NTNU, and the National Unit for Orofacial Pain, Haukeland University Hospital, Faculty of Medicine, University of Bergen.
Participants
Participants were enrolled between May 2018 and February 2021, and the final assessment session took place in November 2021. The flow of participants through the trial is presented in a CONSORT diagram (19) (Figure 1). The clinical diagnosis was based on the ICHD-3 (1,20). The main inclusion criteria were age 18–80 years, a modified version of PIFP criteria (ICHD-3) (Table 1), failed previous treatments and average pain intensity ≥4 (0–10). The modified PIFP criteria (Table 1) specified in addition to the original criteria states that the “patient may denote paresthesia or a mild degree of gain or loss of sensory function in the affected area”. Furthermore, it specified that, in addition to excluding a dental cause, “signs of structural pathology or other specific causes of pain are not identified. Minor operation and injury (insignificant trauma e.g. tooth extraction) to the face, maxilla, teeth and gums without a direct causal relationship with the pain regarding both time and site are accepted. A symptomatic cause of pain has been ruled out by MRI of the brain and facial structures”. More specified details on inclusion and exclusion criteria are provided in Table Supplements 1 and 2. As described in a previous study (3), we employed our modified version of the ICHD-3 criteria because the original criteria are non-specific and difficult to use in practice. There is little empirical evidence validating the original criteria, and we consider these modified criteria to be more suitable (1,20).

CONSORT flowchart shoving inclusion and exclusion/dropouts in different phases of the study.
Modified persistent idiopathic facial pain criteria.

Modification from the diagnostic criteria.
Potentially eligible participants (1, 20) (Table 1) came to a screening visit at the outpatient clinic at the Department of Maxillofacial Surgery at St Olavs Hospital in Trondheim or the National Unit for Orofacial Pain. Baseline characteristics and relevant medical history were registered in a web-based Clinical Record Form (Web-CRF). Patients then completed a 4-week baseline daily PIFP diary on paper, and only those fulfilling the inclusion criteria were randomized to treatment sequence, either A: BTA first, then placebo; or B: Placebo first, then BTA. As described, only participants with an average pain severity >4 were eligible for inclusion; however, this criterion was not revealed to the participants. Those not fulfilling the criteria were considered as screening failures.
After the first injection, there was a 20-week follow-up period. Conceptually, this was considered as a 12-week period where one would most likely see an effect and adverse effects (AEs), in addition to an 8-week washout period. Subsequently, the second injection containing the substance not received in the first injection was followed by a new 12-week period for evaluating potential effects and AEs (Figure 1).
An “end of study” visit was performed by the surgeon following this second 12-week follow-up period. Throughout the entire period from the first injection to the end of the second 12-week follow-up, participants were asked to complete a daily PIFP diary.
During the study, the participants were allowed to use prophylactic medication against PIFP if the dose and frequency had remained unchanged at least 4 weeks before baseline (Table Supplements 1 and 2). All use of medications was recorded. Analgesics used as an acute medication to treat pain attacks were permitted and registered in the pain diary.
Outcome measures
The primary outcome measure was the change from baseline to weeks 5–8 in mean daily pain severity, using a Numeric Rating Scale (NRS) and reported by the participants in the PIFP diary. The 5–8-week period was chosen because earlier pilot studies blocking SPG with BTA showed the best effect in this period (15,16,21). Participants were asked to rate their pain each day on a NRS from 0 (no pain) to 10 (worst imaginable pain) for three different subscales:
NRS mean pain during the last 24 hours (primary outcome measure) NRS of the worst pain during the last 24 hours (secondary outcome measure) NRS of the least pain during the last 24 hours and (secondary outcome measure)
Additionally, participants reported the number of hours with pain >4 during the last 24 hours. To be randomized, participants were required to complete at least 80% of days in the pain diary in the baseline period. After randomization, patients were encouraged to complete the diaries and all participants completed at least 80% of the days.
The secondary outcome measures were changes from baseline to weeks 5–8 in the mean of the highest and lowest reported daily pain severity and the number of hours with moderate to severe pain, the number of pain free days, and the proportion of patients with more than 30% improvement in daily average pain severity weeks 5–8 compared to baseline (“responder rate”). All of these measures were also assessed in weeks 1–4 and 9–12 post-injection in post-hoc analyses. Other secondary outcomes included quality of life measured through different Patient Reported Outcome Measures (PROMs) recorded at baseline and 12 weeks after each injection. This included Chalder fatigue score, 36-Item Short Form Survey (SF-36), McGill Pain Score and Hospital Anxiety and Depression Scale (HADS) (22–26). The primary safety outcome was the overall incidence of AEs and serious adverse events (SAEs). All AEs were followed until resolved or considered stable.
Study procedure
Therapeutic technique
The injections were performed at the Department of Maxillofacial Surgery, St Olavs University Hospital. Navigation-assisted administration of BTA toward the SPG was performed (15,16,21,27). Guided by the MultiGuide®, 25 Allergan-units of Botox suspended in 0.5 ml of isotonic saline or 0.5 ml of placebo (isotonic saline) were injected towards the SPG on the affected side.
Blinding
A computer-generated randomization scheme with blocks of four was used. An independent nurse prepared a syringe with either BTA or placebo based on information in the randomization scheme. The nurse ensured that BTA was fully dissolved and not visible in any way, and then placed the syringe at a pick-up place. To avoid any chance of revealing the content, this nurse was not in direct contact with the participant or any of the study personnel.
Statistical analysis
Sample size determination
A power analysis was not performed as a result of insufficient data on the expected average pain severity and its variability in patients with PIFP. Because this is a rare condition, 30 patients were considered a realistic and pragmatic aim, and the present study can be regarded as a placebo-controlled study to collect efficacy information for power calculations for future randomized controlled trials (RCTs).
Analysis
All statistical analyses were performed using STATA, version 17.0 (StataCorp LLC, College Station, TX, USA). The main null hypothesis (H0) was that the primary outcome would not be different comparing active and placebo treatment. For the primary outcome, a generalized linear mixed model approach was used to estimate the difference in pain under the BTA and placebo periods in weeks 1–4, 5–8 and 9–12 post-injection, with weeks 5–8 prespecified as the timeframe of interest. This method allowed the inclusion of all available daily data, including data from two participants who provided PIFP diary information throughout the first period, but did not receive the second injection. Patient ID was included as a random effect to account for within-subject correlations arising from the cross-over design and repeated measures. The treatment received and timeframe (baseline, 1–4 weeks, 5–8 weeks and 9–12 weeks) were included as categorical fixed effects, along with an interaction term between treatment and the post-injection timeframes. The average change from baseline after BTA and placebo injection and the mean difference between treatments were estimated from the models and are presented together with 95% confidence intervals (CI) and P-values. The secondary outcomes based on the daily PIFP diaries were analysed using the same strategy, including the maximum and minimum pain severity, and the number of hours with moderate to severe pain per day.
The number of participants who achieved a 30% reduction in their average pain severity in each 4-week period was determined by comparing the mean severity for each period to the mean severity in the baseline prior to the first injection. The proportion of responders under the BTA and placebo treatment was compared using a McNemar’s chi-squared test for paired proportions and presented as an odds ratio (OR) with 95% CI and P-values.
Mixed linear models were also used to assess the effect of BTA treatment on the PROMS, with patient ID identified again as a random effect to account for the within-subject correlations between baseline and PROMS measures at 12 weeks after each injection.
In exploratory post-hoc analyses, mixed linear models were also employed to assess the difference between treatments during each week post-injection and to investigate the possibility of a carry-over effect. In the investigation of a possible carry-over effect, the 20 weeks after the first injection and prior to the second injection were analysed comparing sequence arms as a parallel study design.
All participants were included in the intention-to-treat analysis. Safety data are presented for all patients who had received at least one treatment.
The study was blinded until the statistical evaluations were finished for all participants of the study group, including the principal investigator/project manager, co-authors and statistician.
Ethical statement
All subjects provided their written informed consent and the study was monitored by the Unit for Applied Clinical Research. The study was approved by the Regional Committee for Medical and Health Research Ethics (REK 2017/1767) and the Norwegian Medicines Agency (EUDRACT number: 2017-002518-30). The study protocol is registered on ClinicalTrial.gov (NCT03462290). Investigations were conducted in accordance with the Declaration of Helsinki, as well as guidelines for good clinical practice (Good Clinical Practice, CPMPIICH/135/95). The reporting conforms to the Consort 2010(19).
Results
During the recruiting period, 60 patients were assessed for eligibility, and 41 participants were included and completed the baseline period. Ultimately, 30 participants were randomized and received at least one treatment. Fifteen participants were assigned to treatment sequence A and 15 were assigned to treatment sequence B. Two subjects dropped out after the first injection (one in each sequence) and one after the second injection (in sequence B) (Figure 1). In total, 29 participants provided headache diary data for at least the first treatment period and 27 participants completed the entire trial. All participants completed more than 80% of days in the PIFP diary when they were active in the study, and none had changed the dose or frequency of preventive treatment. The patients were long-standing sufferers, with both stable symptoms and medications for PIFP. We did not register any changes in preventive or symptomatic medications for PIFP during the study. During the trial, the following medications were used (number of participants using the stated medication); paracetamol (n = 15), tramadol (n = 3), ibuprofen (n = 2), baclofen (n = 1), oxycodon (n = 1), tapentadol (n = 1) and amitriptyline (n = 1). On the whole, baseline characteristics were similar in the two groups at entry (Table 2). More than 10 years of pain was reported by 44% of the participants. The baseline mean for average pain intensity on the NRS scale was 6.21 (95% CI = 5.91–6.52).
Baseline demographics and clinical characteristics by sequence and by total.

Data are n (%) unless stated otherwise.
Includes two subjects who received study drug or placebo in the first intervention but were not randomized to the second intervention and one subject who received study drug or placebo in the second intervention but dropped out after the second injection. One patient completed the hole study but with missing demographic and questionnaire data from baseline. PIFP: persistent idiopathic facial pain.
Outcome measures
Average pain severity (primary outcome) showed no difference between BTA and placebo in weeks 5–8 (mean difference = 0.00, 95% CI = –0.57 to 0.57) (Table 3). The difference between the BTA and placebo treatment for the other measures of pain severity and the number of hours with moderate to severe pain was observed to be small and not statistically significant during weeks 5–8 after the injection (Figure 2, Table 3). In total, five patients experienced a reduction of at least 30% in their average pain severity from baseline to weeks 5–8 in each of the BTA and the placebo treatment periods (Table 4). Similarly, under each treatment period, 4 participants reported an average pain score <4. No participants experienced pain free days during weeks 5–8 post-injection. For the quality of life measures, there was no apparent difference between the two treatments (Table 5).
Pain severity outcomes and number of hours in BTA versus placebo group at baseline and at weeks 5–8.

BTA, onabotulinum toxin A; CI, confidence interval. The mean (*), mean differences (**) and 95% CIs were estimated from the mixed linear regression models including 29 patients who had completed at least one period. Two participants dropped out before period two, both of whom had received only the BTA treatment.

Primary outcome: change from baseline to weeks 1–12 in NRS comparing BTA and placebo. Means and 95% CIs of primary outcome measures are shown for each group at each time period. (Colour: red = placebo, blue = BTA). Y = NRS, X = weeks. Weeks 5–8 are the primary endpoint measure. NRS, numeric rating scale; BTA, onabotulinum toxin A.
Number of participants with a 30% reduction in average pain severity from baseline (n = 27).

27 participants included in analyses (13 in BTA; 14 in placebo).
Post-hoc analyses
Post-hoc analyses included estimation of the BTA treatment effect on pain severity, number of hours and proportion of participants with at least 30% response in the periods weeks 1–4 and weeks 9–12 post-injection (Table 4; see also Table Supplement 3).
During weeks 1–4 post-injection, there was a statistically significant improvement in average pain, worst pain, and number of hours with moderate to severe pain in favour of BTA (Figure 3; see also Table Supplement 3). Five participants had at least a 30% reduction in average pain during the first 4 weeks of active treatment and only two following placebo, although this difference did not reach statistical significance (Table 4). Also during weeks 1–4 post-injection, there were six participants in the BTA group with an average pain score <4 and only one participant in the placebo group.
In weeks 9–12, there was no observed difference in average pain, worst pain or number of hours with moderate to severe pain comparing BTA to placebo (Figure 2; see also Table Supplement 3). There were seven participants with more than 30% reduction in the average pain after BTA and four participants after placebo (Table 4). Also during weeks 9–12 post-injection, there were six and four participants with an average pain score <4 when under the BTA and placebo treatment period, respectively.
Exploring a potential carry-over effect, we analysed the two sequence arms as if they had been a parallel study up until the second injection. The group who received BTA at the first injection had a lower average pain throughout the whole first period, with statistically significant differences seen in weeks 1–4, weeks 13–16 and weeks 17–20 compared to the group who received placebo first (Figure 3; see also Table Supplement 4). The reduction in pain intensity from baseline after BTA injection was greater for the group receiving the active injection in the first period compared to those participants who first received BTA in the second treatment period, although the difference in change from baseline was not statistically significant during any 4-week period (Table Supplement 5).

Primary outcome demonstrated by treatment sequence A and B. Means and 95% CIs of outcome measures for each treatment sequence are shown at each time period. Colour: green = sequence A (BTA/placebo), red = sequence B (placebo/BTA). Y = NRS, X = weeks. Weeks 5–8 are the primary endpoint measure. NRS, numeric rating scale.
When considering all 20 weeks following the first injection, participants who had received BTA had a lower overall average daily pain compared to those who had received placebo, with a mean difference in average daily pain in weeks 1–20 of –0.94 (95% CI = −1.84 to –0.04, P = 0.040). In the placebo group, we observed a 3.8% reduction in average pain intensity over the first 20 weeks and a 20.6% reduction after BTA injection, which is a difference of 16.8 percentage points.
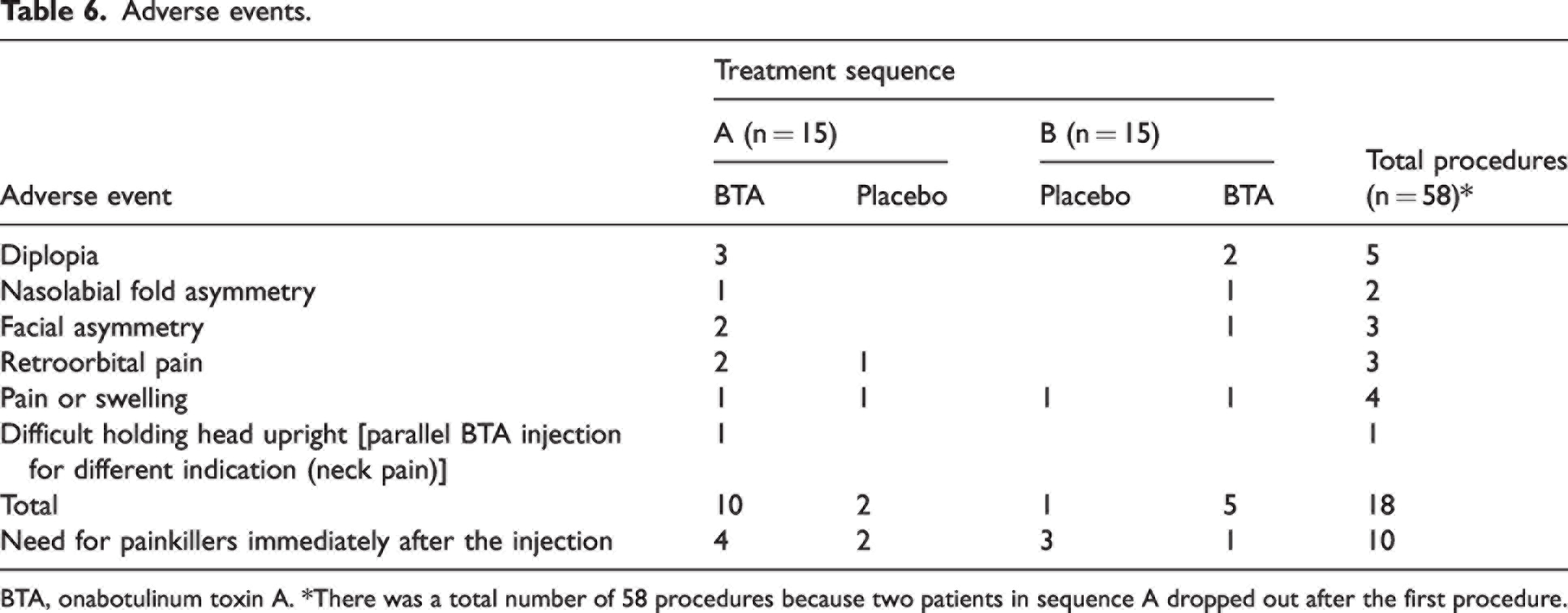
Adverse events
No SAEs were reported. Twelve out of 30 participants experienced AEs (Table 6). AEs were predominantly mild and did not require any treatment. Five participants experienced diplopia, of which two lasted for hours to days, and three lasted for 1–3 months and were considered bothersome. The three participants who experienced diplopia also experienced facial asymmetry. Ten participants had to take additional analgesics on the day of the injection. Of 18 AEs observed, nine were considered device-related AEs and 10 were considered study-drug related. One related to BTA but not related to the study drug because the participant received a non-study BTA injection for neck pain. All AEs resolved within 14 weeks after injection. The mean (SD) pain at the injection site was 2.8 (2.42) graded by the participants on an NRS scale (0–10) post injection.
Summary of patient reported outcome measures (PROMs).
PROMs reported at baseline and 12 weeks post intervention by BTA and placebo. Mean and 95% CI. Sequence A (BTA/placebo), sequence B (pla cebo/BTA).
Adverse events.

BTA, onabotulinum toxin A. *There was a total number of 58 procedures because two patients in sequence A dropped out after the first procedure.
Discussion
This is the first RCT with BTA treatment in PIFP patients. There was no apparent difference between BTA and placebo in the average pain during weeks 5–8 post-injection. Similarly, BTA did not appear to affect the maximum or minimum pain level experience, nor the number of hours with moderate to severe pain in weeks 5–8 post-injection or quality of life 12 weeks after injection. However, average and maximum pain intensity, as well as duration of pain, were significantly lower during the first 4 weeks after BTA injection. Although the conceptual washout length period in the present study was based on previous studies considering the BTA effect on migraine (PREEMPT) (28), further analyses suggested there may have still been a carry over effect that could have underestimated the effect of BTA.
In post-hoc analyses comparing treatments during the first period only as a parallel design, we found that the pain reduction persisted beyond the expected period in sequence A (BTA/placebo), through the conceptual washout period and potentially affected period 2 (Figure 3). As shown in Figure 3, the placebo group returns to the baseline level at the end of the washout period, whereas the BTA group does not. This demonstrates a possible carry-over effect in participants randomized to sequence A (BTA/placebo) and a subsequent underestimation of their pain reduction after BTA compared to placebo because their pain levels during the placebo period are likely to be still influenced by the previous BTA injection. We also observed that the reduction in pain intensity compared to baseline following BTA injection was greater among those who received the active treatment during the first period (sequence A) compared to those who first had the BTA injection in the second period (sequence B). This difference in change from baseline was not statistically significant, and it is not possible to determine whether this was a result of random variation or a difference between the groups, or even a result of having undergone the placebo injection first.
The post-hoc analyses also suggest that pain relief lasted 20 weeks in sequence A (BTA/placebo), which is longer than expected based on previous trials targeting the SPG for migraine prophylaxis (15,16,21) where the most pronounced effect was evident during weeks 5–8. With a mean reduction in their average pain of 16.8%, the overall effect seen during the 20 weeks following the first injection was not negligible. It has been suggested that BTA may have an additional regulatory role in addition to the synaptic blockage of parasympathetic signals in the SPG and this may provide an explanation for some of the persistent effects seen in the present study compared to other studies (13).
Surprisingly, the prevalence of depression and anxiety in subscales, as well as total HADS, was low in the quality of life questionnaires. This contradicts previous findings regarding PIFP patients (29). We consider that this may partly result from the stringent criteria for admission to the study, excluding patients with severe psychiatric conditions. Overall, our impression was that psychiatric comorbidity was not very prominent among patients considered for the present study.
Altogether, the present study has demonstrated that the intervention was safe and well-tolerated for patients with PIFP, substantiated by the high number of participants finishing the 32-week trial (90.0%). AEs in the present study were comparable to those in previous studies with the same navigational tool (MultiGuide®) and procedures (15, 16, 21). Transient diplopia is likely caused by paresis of the musculus rectus inferior and has also been seen in other studies with the same technique (30).
As a result of the experimental nature of the treatment, only the most refractory and severely affected patients were included in the present study. The included participants were characterized by long disease duration, long-lasting facial pain measured in hours of facial pain per day and high average pain intensity. This may also explain the late commencement of the effect in this population because the persistent and long-lasting pain may have delayed a putative effect of BTA on central sensitivity. One might expect a greater effect in patients with a shorter pain history and there is reason to hope that there would be a greater effect in these patients.
Currently, few alternatives with adequate scientific documentation exist for treating PIFP (4). One trial targeting SPG with alcohol (14) reported a ≥50% pain reduction lasting ≥1 month, but with a recurrence rate of 72.3% after a mean duration of 5.4 months. Another recent cross-over study showed that high-frequency repetitive transcranial magnetic stimulation could alleviate treatment-resistant chronic facial pain (31).
The present procedure may be comparable in terms of efficacy and duration of pain relief to previously described procedures. However, direct comparisons of the outcomes should be considered cautiously as a result of differences in craniofacial pain conditions studied, inclusion criteria, procedures and definitions of treatment success. The present study indicates that BTA injection towards the SPG by the MultiGuide® is a feasible approach. In particular, this technique does not require the sophisticated devices that are needed for stimulation or radiofrequency, in addition to being a minimally invasive procedure compared to surgical treatments, with low complication rates.
Therefore, in view of the scarcity of therapeutic options, and the high degree of suffering of these patients, we find the results of the present study to be encouraging. The post-hoc analyses, considering the study as a parallel group study after the first injection to avoid the potential carry-over effect, provided evidence of an effect that started within the first 4 weeks and lasted for at least 20 weeks. A new study should be designed as an adequately powered parallel group study. Future studies should also consider the use of two or three injection cycles, aiming to determine whether a potential effect increases over time with more injections, as well as possibly an open extension period, aiming to evaluate effect duration and allow the collection of long-term safety data. Because the intervention has been safely administered in this and other studies (15,16,21), the inclusion of less severely affected patients could be considered and, ultimately, may be a treatment option for patients not considered “treatment-resistant”.
Limitations
The main limitation of the present study was the cross-over design with a carry-over effect and, consequently, a potential efficacy may have been lost. Another limitation of the present study is the small number of participants. As a result of insufficient data on the expected average pain severity and its variability in patients with PIFP, a power analysis was not performed. Given the rarity of this condition, 30 patients were deemed a realistic and pragmatic target, and the present study can be viewed as a placebo-controlled study to acquire efficacy data for future RCT power calculations. Some AE, such as facial asymmetries and diplopia, may have unblinded these participants. Future studies should attempt to reduce such AEs by optimizing the technique. Regarding the lack of effect on PROMs, it may be that a significant change was not detected in the present study because the PROMs were assessed some time after and not during the time period of a potential BTA effect. In general, cross-over studies can be affected by a high drop-out rate, although this was not the case in the present study.
Conclusions
Injection of BTA toward the SPG with the MultiGuide® did not appear to provide a pain reduction at 5–8 weeks in patients with chronic PIFP. Post-hoc analyses suggest that the effect of BTA may arise already in the first 4 weeks post-injection, and an apparent carry-over effect may have led to an underestimation of the effect of BTA at later time points in the cross-over design. Therefore, an important learning point for future studies is that an adequately powered parallel study would be preferable. The injection otherwise appears to be safe and well-tolerated in patients with PIFP.
Clinical implications
Injection of BTA toward the SPG with the MultiGuide® did not appear to provide a reduction in pain reduction at 5–8 weeks in patients with chronic PIFP. Post-hoc analyses suggest that the effect of BTA may arise already in the first 4 weeks post-injection There is some indication that this cross-over study design has masked the effect of BTA The injection appears to be safe and well-tolerated in patients with PIFP Learning point. For future studies, it would be advisable to conduct a parallel study or apply a significantly longer washout period
Supplemental Material
sj-pdf-1-cep-10.1177_03331024231187132 - Supplemental material for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study
Supplemental material, sj-pdf-1-cep-10.1177_03331024231187132 for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study by Kent A. Jamtøy, Wenche M. Thorstensen, Lars J. Stovner, Annika Rosén, Stine Maarbjerg, Daniel Bratbak, Melanie R. Simpson and Erling Tronvik in Cephalalgia
Supplemental Material
sj-pdf-2-cep-10.1177_03331024231187132 - Supplemental material for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study
Supplemental material, sj-pdf-2-cep-10.1177_03331024231187132 for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study by Kent A. Jamtøy, Wenche M. Thorstensen, Lars J. Stovner, Annika Rosén, Stine Maarbjerg, Daniel Bratbak, Melanie R. Simpson and Erling Tronvik in Cephalalgia
Supplemental Material
sj-pdf-3-cep-10.1177_03331024231187132 - Supplemental material for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study
Supplemental material, sj-pdf-3-cep-10.1177_03331024231187132 for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study by Kent A. Jamtøy, Wenche M. Thorstensen, Lars J. Stovner, Annika Rosén, Stine Maarbjerg, Daniel Bratbak, Melanie R. Simpson and Erling Tronvik in Cephalalgia
Supplemental Material
sj-pdf-4-cep-10.1177_03331024231187132 - Supplemental material for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study
Supplemental material, sj-pdf-4-cep-10.1177_03331024231187132 for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study by Kent A. Jamtøy, Wenche M. Thorstensen, Lars J. Stovner, Annika Rosén, Stine Maarbjerg, Daniel Bratbak, Melanie R. Simpson and Erling Tronvik in Cephalalgia
Supplemental Material
sj-pdf-5-cep-10.1177_03331024231187132 - Supplemental material for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study
Supplemental material, sj-pdf-5-cep-10.1177_03331024231187132 for Onabotulinum toxin A block of the sphenopalatine ganglion in patients with persistent idiopathic facial pain: a randomized, triple-blind, placebo-controlled, exploratory, cross-over study by Kent A. Jamtøy, Wenche M. Thorstensen, Lars J. Stovner, Annika Rosén, Stine Maarbjerg, Daniel Bratbak, Melanie R. Simpson and Erling Tronvik in Cephalalgia
Footnotes
Acknowledgements
We thank all the participants for taking part in this study. We also thank Kristin Elden for coordinating the study; Irina Aschehoug, Gry Altmann Krogstad and May Karin Ness for the surgical assistance and blinding procedure; and the Department of Maxillofacial Surgery at St Olavs University Hospital, Norway, for facilitating the clinical study.
Author contributions
KAJ and ET had the original idea for the manuscript and LJS and SM contributed to the initial planning phase. KAJ and MRS analysed the data. KAJ reviewed the literature for the introduction and discussion and drafted the first version of the manuscript. WMT, LJS, AR, SM, DB, MRS and ET assisted in the interpretation of the results, and drafting and revision of the manuscript. All authors read and approved the final version of the manuscript submitted for publication.
Declaration of conflicting interests
Professor Tronvik and Dr Bratbak may benefit financially from a commercialization of a proposed treatment targeting the SPG and the intervention device used to perform the treatment through future possible intellectual properties. Dr Jamtøy, Dr Thorstensen, Professor Stovner, Professor Rosén, Dr Maarbjerg and Dr Simpson declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by an innovation grant given by NTNU (Norwegian University of Science and Technology)
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.