
Abstract
Background
We performed a random-effects network meta-analysis to study the efficacy and safety of newly developed drugs for the acute treatment of migraine attacks.
Methods
MEDLINE via PubMed, Embase and The Cochrane Register of Controlled Trials were searched from inception to 11 February 2022. Phase 3 randomized controlled trials examining all formulations of lasmiditan, rimegepant and ubrogepant for the acute treatment of adults with migraine, were included. Data were extracted following the PRISMA guidelines.
Results
Seven studies (SAMURAI, SPARTAN, CENTURION, Study 302, Study 303, ACHIEVE I and II) involving n = 12,859 patients were included. All treatments were superior in efficacy to placebo. Lasmiditan 200 mg showed the highest two-hour pain freedom, while two-hour freedom from most bothersome symptom was equally achieved by the higher doses of lasmiditan (100 and 200 mg), rimegepant and the higher doses of ubrogepant (50 and 100 mg). The odds of treatment-emergent adverse events were greatest with all doses of lasmiditan.
Conclusion
Lasmiditan 200 mg was the most effective intervention in the treatment of migraine attacks, although it was associated with high degrees of dizziness, nausea and somnolence. Rimegepant showed slightly lower, but similar efficacy rates to lasmiditan. Ubrogepant had overall the best tolerability profile. These conclusions are limited by the absence of head-to-head comparisons, limitations of individual trials and of the meta-analysis methodology itself.
Introduction
Migraine is currently recognized as one of the leading causes of disability worldwide and a major public health issue (1). The recent years have seen a dramatic expansion in options for acute migraine management, particularly with the discovery and large-scale testing of two new generations of treatments: ditans and gepants (2,3). These drugs possess the significant advantage of having been developed and designed specifically for the abortion of the migraine attacks. Like the widely used triptans, they are able to target migraine-specific pathways, without the caveat of vasoconstriction that reduces the bandwidth of triptan use.
Ditans, potent and selective of the 5-HT1F receptor agonists, are represented by lasmiditan. This is available orally at the doses of 50, 100 and 200 mg and has been approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the acute treatment of migraine with and without aura in adults (4,5).
Gepants are small molecule calcitonin gene-related peptide (CGRP) receptor antagonists, available in oral formulations and with a nasal formulation in development (6). The first gepants to be tested in humans were either unavailable in oral formulation (olcegepant [7]), caused liver toxicity (telcagepant [8] and MK-3207 [9]) or were not pursued for commercial reasons (10). Rimegepant and ubrogepant represent a new generation of oral gepants for acute migraine therapy, which have shown no hepatotoxicity in phase 2 clinical trials (11,12). Rimegepant is available at the dose of 75 mg in orally disintegrating form. Ubrogepant was studied at the doses of 25, 50 and 100 mg, however, it has only been approved commercially in the 50 and 100 mg formulations.
Overall, clinical data for these drugs is very recent, with most clinical trials published in the last few years.
With this systematic review and network meta-analysis of phase 3 randomized controlled trials (RCTs) we aim to examine and compare the efficacy and safety of all formulations of lasmiditan, rimegepant and ubrogepant for the acute treatment of migraine in adult patients.
Methods
Study design
This systematic review and meta-analysis followed the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) and was registered in the International Prospective Register of Systematic Reviews (PROSPERO: CRD42022308224). A team of four clinical headache scientists (FP, SY, ML, FH) defined the inclusion and exclusion criteria as well as the primary and secondary outcome measures, executed the search strategy, selected relevant publications, and performed data extraction and risk of bias assessment as described below. Statistical analysis was conducted by a bio-statistician (EMH). All steps were reviewed by senior authors PJG and CT.
Search strategy and study selection
MEDLINE via Pubmed, EMBASE and Cochrane central register of controlled trials were searched from inception until 11 February 2022. The detailed search strategy is provided in the online Supplementary Table 1. The PRISMA flowchart of screened studies is shown in Figure 1. Following the initial search, two pairs of independent reviewers (FP, SY, ML, FH) performed the following steps: deletion of duplicates, screening of titles and abstracts of all citations, and retrieval of papers for full-text screening. Disagreements were solved by discussion, and if necessary by a third senior author (PJG, CT). The Rayyan platform for systematic reviews (http://rayyan.qcri.org) was used for study selection and screening (13).

PRISMA diagram of screened studies.
Following our PICOS criteria (online Supplementary Table 2), studies were included if they: 1) were phase 3 double-blind RCTs examining relevant primary and secondary outcomes for adult episodic and chronic migraine patients, diagnosed according to the International Classification of Headache Disorders criteria, Third edition (ICHD-III) (14) or the ICHD operating at the time of the study; 2) included lasmiditan oral (50, 100, 200 mg), rimegepant oral (75 mg) or ubrogepant oral (25, 50, 100 mg), as intervention; 3) compared intervention to placebo; 4) were fully published in English language. Exclusion criteria were: studies not including adult patients, studies not published in English, studies not available as full papers (e.g. conference abstracts), phase 1 or 2 studies, post-hoc or secondary analyses, studies not comparing one of the intervention measures (i.e. lasmiditan, rimegepant and ubrogepant) to placebo.
Primary and secondary outcomes measures
All outcomes for efficacy and safety followed the guidelines of the International Headache Society (IHS) for controlled trials of acute treatment of migraine attacks in adults (15). The two primary outcomes for efficacy were: pain freedom at two hours before the use of any rescue medication and freedom from most bothersome symptom (MBS) associated with the treated migraine attack at two hours. Secondary efficacy outcome measures were: pain relief at two hours, sustained pain freedom (percentage of subjects who are pain free at two hours with no use of rescue medication nor relapse, within 24 or 48 hours of the initial treatment), sustained pain relief at 24 and 48 hours, freedom from photophobia at two hours, freedom from phonophobia at two hours and freedom from nausea at two hours.
The main outcomes for safety and tolerability were: any recorded adverse event (AE) within 48 hours post dose (also termed treatment emergent adverse events, TEAEs), any serious adverse events (SAEs), number of patients who discontinued treatment due to AEs and frequency of most common TEAEs (i.e. nausea, dizziness, paresthesia, somnolence, fatigue, lethargy, palpitations, dry mouth, urinary tract infections).
Data extraction and quality assessment
A predesigned excel spreadsheet was used for data extraction, which included: title of the study, name of first author, year of publication, demographics of study population (age, gender, ethnicity), sample size, interventions, duration of intervention, baseline data, outcomes and information for assessment of the risk of bias. Any conflict in data extraction was resolved by discussion or by a third reviewer (FP, SY, ML, FH). The Revised Cochrane risk-of-bias tool for randomized trials tool was used for assessing risk of bias (RoB 2) (16), performed by two reviewers independently (FP, FH) using the methodology described in Part 2, Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (17). Specifically, RoB 2 is structured into a fixed set of domains of bias, focusing on different aspects of trial design, conduct and reporting, including: bias arising from the randomization process, bias due to deviations from intended interventions, bias due to missing outcome data, bias in measurement of the outcome and bias in selection of the reported result. The overall risk of bias is reported as the least favorable assessment across these five domains.
Statistical analysis
A frequentist random effects network meta-analysis was applied to the primary and secondary outcome measures (18,19). In addition, pairwise meta-analyses were performed using the inverse variance method with restricted maximum likelihood (REML) estimation (20). Odds ratios with 95% confidence intervals were estimated for all outcomes. All treatments were ranked based on primary and secondary outcomes with P scores, a frequentist analogue to the Surface Under the Cumulative RAnking curve (SUCRA) with a similar interpretation (21). All analyses were performed in R version 4.1.2 (22). Heterogeneity is here analyzed for the pairwise comparisons in a graphical way. In addition, the I2 index, the between study variance tau2 as well as the P value for Cochrane’s Q test are presented.
Results
Search results
A total of 712 records were identified from the literature search (Figure 1). After removal of 362 duplicates, 350 papers were screened through title and abstract, of which 336 were further removed. Full-text screening was performed on 14 papers; two were excluded because they were conference abstracts, while four were excluded because they were not phase 3 RCTs. Seven studies fulfilling the inclusion criteria were included in the final network meta-analysis.
Included studies
All included studies were published between 2018 and 2021 and took place in the USA, Europe and parts of Asia (Table 1). The studies collectively involved a total of n = 12,859 randomized patients. Three studies examined lasmiditan (n = 6849): SAMURAI (23), SPARTAN (24) and CENTURION (25); two ubrogepant (n = 3358): ACHIEVE I (26) and ACHIEVE II (27) and two rimegepant (n = 2652): Study 302 for the oral tablet (28) and Study 303 for the orally disintegrating tablet (ODT) (29).
Baseline characteristics of included studies.

ITT, intention to treat; mITT, modified intention to treat; ODT, orally disintegrating tablet; -: not reported.
Patients in CENTURION used lasmiditan across four different migraine attacks. In the remaining studies, patients used the intervention for a single migraine attack.
Participant demographics and baseline headache features were balanced across studies. Risk of bias assessment showed low risk on all seven studies across the five domains (online Supplementary Figure 1). A network of the included interventions is shown in Figure 2.
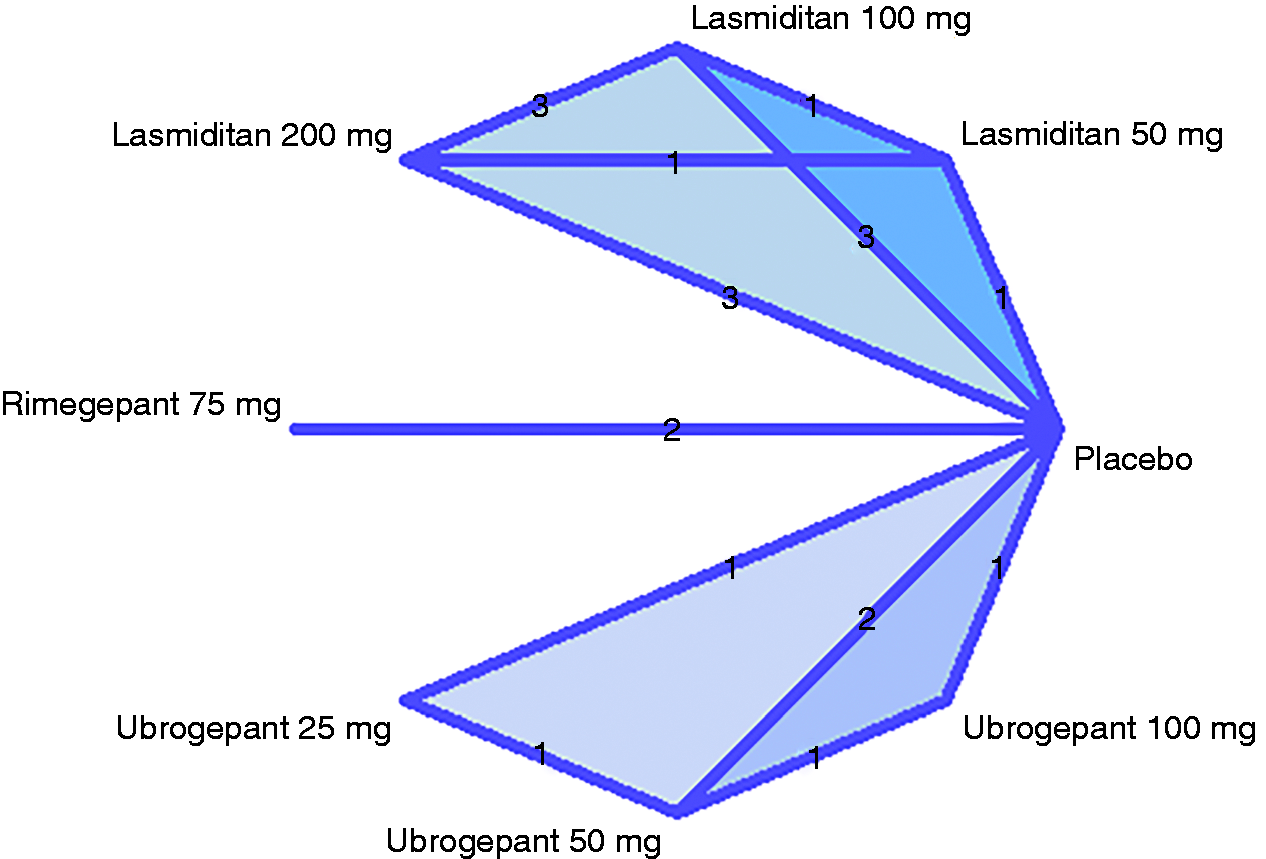
Map showing network of treatments and number of included studies for each intervention.
All studies excluded patients with chronic migraine, medication overuse headache and individuals with cardiovascular risk factors. An exception for the latter were: SPARTAN, which did not exclude individuals with known coronary artery disease, clinically significant arrhythmia, or uncontrolled hypertension; and CENTURION, in which individuals with known cardiovascular risk factors or disease, with the exception of history of hemorrhagic stroke, were included.
Efficacy outcomes
The network meta-analysis for the primary efficacy outcome of two hours pain freedom showed that all interventions were significantly superior to placebo (Figure 3a), with lasmiditan 200 mg showing the highest rates (OR 2.88 [95% CI: 2.22; 3.73]), and ubrogepant 25 mg showing the lowest (OR 1.59 [95% CI: 1.03; 2.47]). The second most effective treatment was lasmiditan 100 mg (OR 2.28 [95% CI: 1.75; 2.96]), followed by rimegepant and ubrogepant 100 mg, which showed similar efficacy (OR 2.0 [95% CI: 1.45; 2.75] and OR 1.97 [95% CI: 1.27; 3.07], respectively). All treatments were superior to placebo regarding freedom from MBS at two hours. Lasmiditan 200 mg (OR 1.66 [95% CI: 1.42; 1.93]) and ubrogepant 50 mg (OR 1.66 [95% CI: 1.36; 2.02]) showed the highest efficacy with the same odds ratio.

Forest plots for primary (a) and secondary (b) efficacy outcomes. Results to the left of 1 favor placebo, to the right favor intervention.
The second most effective treatments for MBS at two hours were rimegepant 75 mg (OR 1.61 [95% CI: 1.35; 1.91]), lasmiditan 100 mg (OR 1.61[1.38; 1.87]) and ubrogepant 100 mg (OR 1.60 [95% CI: 1.23; 2.07]), with similar odds ratios. Lasmiditan 50 mg (OR 1.3 [95% CI: 1.04; 1.62]) and ubrogepant 25 mg (OR 1.36 [95% CI: 1.04; 1.77]) showed the lowest efficacy for this outcome.
Comparison of the two primary efficacy outcomes between all interventions (Table 2) supported the results presented in Figure 3a. The comparison analysis showed that lasmiditan 200 mg was estimated significantly superior to the 50 mg dose of the same drug for two-hour pain freedom (OR 1.64 [95% CI: 1.14; 2.35]) and freedom from MBS (OR 1.28 [95% CI: 1.03; 1.59]). Ubrogepant 50 mg showed numerically equal efficacy to lasmiditan 200 mg for freedom from MBS (OR 1 [95% CI: 0.78; 1.28]).
Comparisons between all interventions for the two primary efficacy outcomes shown as OR [95% CI]. The upper (light grey) triangle shows comparisons (column vs row) for two-hour pain freedom; the lower (dark grey) triangle shows comparisons (row vs column) for two-hour freedom from most bothersome symptom. ORs higher than 1 in the upper triangle favor the column-defining drug; ORs higher than 1 in the lower triangle favor the row-defining drug.

Among secondary outcome measures (Figure 3b), freedom from photophobia at two hours was significantly achieved for all interventions compared to placebo except for ubrogepant 25 mg. All treatments provided higher odds of freedom from phonophobia at two hours compared to placebo. Freedom from nausea at two hours was only achieved for rimegepant (OR 1.25 [95% CI: 1.02; 1.53]) and ubrogepant 50 mg (OR 1.24 [95% CI: 1.01; 1.51]) with respect to placebo. Ubrogepant 100 mg presented the highest odds ratio for freedom from nausea at two hours, which was not statistically significant (OR 1.27 [95% CI: 0.98; 1.65]), while lasmiditan 50 mg performed worse than placebo (OR 0.94 [95% CI: 0.73; 1.22]).
Sustained pain freedom at 24 hours was achieved for all treatments compared to placebo, except for ubrogepant 25 mg (OR 1.54 [95% CI: 0.87; 2.74]). Pain relief at two hours was significantly achieved for all treatments. Sustained pain relief at 24 hours (not measured in the lasmiditan studies), showed more than twice as high odds ratios as in the placebo group for rimegepant (OR 2.24 [95% CI: 1.89; 2.65]) and ubrogepant 50 (OR 2.18 [95% CI: 1.76; 2.7]) and 100 mg (OR 2.34 [95% CI: 1.78; 3.08]).
Data for primary outcomes were available for all studies. Freedom from photophobia, phonophobia and nausea at two hours were not reported in the CENTURION study (lasmiditan 100, 200 mg). Pain freedom and sustained pain relief at 48 hours were not reported in the ubrogepant studies. Sustained pain relief was not reported for any time point in the lasmiditan studies. We were unable to perform the analyses for secondary outcomes sustained pain relief and pain freedom at 48 hours due to lack of data.
Pairwise comparisons for each intervention were performed in addition to the network meta-analysis and are reported in Figure 4 for the two main efficacy outcomes, and in the supplementary material for remaining outcomes. Although the results of this analysis are largely comparable, some discrepancy can be seen for the two-hour pain freedom outcome. Here, the estimated primary treatment effect of 100 and 200 mg lasmiditan in CENTURION with respect to SAMURAI and SPARTAN shows a large heterogeneity across studies. For the remaining outcomes results were mostly homogeneous.

Pairwise comparisons for primary efficacy outcomes.
Safety and tolerability outcomes
Odds of treatment-emergent adverse events were significantly higher with lasmiditan than placebo (Figure 5); particularly the 200 mg dose (OR 4.62 [95% CI: 3.73; 5.72]) showed the largest effect. TEAEs were also slightly more likely with ubrogepant 100 mg (OR 1.52 [95% CI: 1.02; 2.29]) and rimegepant (OR 1.29 [95% CI: 0.94; 1.77]), although this was not a significant finding for rimegepant. On the other hand, TEAEs were less likely to occur with ubrogepant 25 mg than with placebo (OR 0.76 [95% CI: 0.48; 1.2]) and almost equally likely with ubrogepant 50 mg (OR 0.97 [95% CI: 0.7; 1.35]).

Forest plots for main safety and tolerability outcomes: frequency of any TEAE, nausea, dizziness, paresthesia and somnolence. Results to the left of 1 favor placebo, to the right favor intervention.
Nausea was estimated more likely than placebo with all interventions, except for ubrogepant 25 (OR 1.26 [95% CI: 0.57; 2.8]). and 50 mg (OR 1.03 [95% CI: 0.53; 1.99]). Notably, nausea showed the highest odds with lasmiditan 200 mg (OR 2.69 [95% CI: 1.84; 3.93]) and ubrogepant 100 mg (OR 2.55 [95% CI: 1.21; 5.37]).
Dizziness was not reported in ACHIEVE I and so data are not available for ubrogepant 100 mg, however this side effect was less likely with the 50 mg ubrogepant dose (OR 0.89 [95% CI: 0.31; 2.53]) and rimegepant 75 mg (OR 0.87 [95% CI: 0.29; 2.65]), than with placebo. On the other hand, lasmiditan was linked with very high odds of developing dizziness compared to placebo, particularly at higher doses (for 200 mg, OR 6.96 [95% CI: 5.12; 9.47]; for 100 mg, OR 5.88 [95% CI: 4.31; 8.03]).
The odds ratio of a somnolence event (not reported in the rimegepant studies and ACHIEVE II) was estimated as very large, with values above 2.5 with all lasmiditan doses (for 200 mg, OR 3.48 [95% CI: 2.28; 5.33]; for 100 mg, OR 2.57 [95% CI: 1.66; 3.97]; for 50 mg, OR 2.89 [95% CI: 1.64; 5.1) as well as ubrogepant 100 mg (OR 3.05 [95% CI: 0.94; 9.85]) compared to placebo, whereas the odds was estimated smaller with ubrogepant 50 mg than with placebo (OR 0.78 [95% CI: 0.17; 3.59]).
Fatigue and paresthesia were reported in the lasmiditan studies only and were estimated higher than placebo with all doses, except for paresthesia with lasmiditan 50 mg which did not reach statistical significance.
For the adverse events of palpitations, lethargy, urinary tract infections (UTIs) and dry mouth not enough data were present to justify meta-analyses. Palpitations were reported in the lasmiditan studies only and were present in n = 4 patients in the placebo groups (0.2% of total aggregated cohort), in n = 2 (0.3%) for lasmiditan 50 mg, n = 16 (0.9%) for lasmiditan 100 mg and n = 13 (0.8%) for the lasmiditan 200 mg groups.
Lethargy was reported in SAMURAI and SPARTAN in a total of n = 3 patients (0.2%) in the placebo groups, n = 8 patients (1.2%) in the lasmiditan 50 mg groups, n = 20 (1.6%) in lasmiditan 100 mg groups and n = 29 (2.3%) in patients taking lasmiditan 200 mg.
UTIs were only reported in the rimegepant studies, present in n = 18 patients (1.5%) taking rimegepant 75 mg and n = 10 patients (0.8%) taking placebo.
Dry mouth was reported only in CENTURION and ACHIEVE I for a total of n = 3 patients (0.3%) in the placebo groups, n = 6 (1.2%) in the lasmiditan 100 mg group, n = 11 (2.3%) in the lasmiditan 200 mg group, n = 3 (0.6%) in the ubrogepant 50 mg group and n = 10 (2.1%) in the ubrogepant 100 mg group.
No serious adverse events were recorded in Study 303 (rimegepant 75 mg ODT), ACHIEVE I and II (ubrogepant). In the CENTURION study, five serious adverse events were considered to be treatment-related: two patients in the placebo group developed liver disorder and suicidal ideation, one in the lasmiditan 100 mg group had asthma, and two in the lasmiditan 200 mg reported hemiplegic migraine and serotonin syndrome. In SAMURAI, two patients on lasmiditan 200 mg and one in the placebo group reported serious TEAEs, although these were not thought to be related to the study drug. In the SPARTAN study, a total of five serious adverse events were reported, of which two were considered related to the treatment: a dystonic reaction in the 100 mg group and a presyncope in the 200 mg group. Both events resolved with sequelae (a positive Romberg test and fatigue, respectively). In Study 302, serious adverse events were reported for one patient in the rimegepant group (back pain) and for two patients in the placebo group.
In CENTURION, 36 participants on lasmiditan 100 mg and 38 on lasmiditan 200 mg withdrew because of adverse events related to the study drug, while six patients withdrew in the placebo group. One study participant in the 200 mg lasmiditan group in SPARTAN study discontinued because of fatigue and dizziness. No other subjects in the remaining studies withdrew after taking the study medication. Withdrawals were not reported for Study 302 (rimegepant 75 mg).
Treatment rankings
When ranking all treatments for the available outcomes, lasmiditan 200 mg shows the best performance for two-hour pain freedom as well as most secondary efficacy outcomes (Table 3). However, for two-hour MBS, lasmiditan 200 mg (75%) ranked almost equally to ubrogepant 50 mg (74%), confirming the overall results of the meta-analysis. Importantly, lasmiditan 100 and 200 mg ranked lowest on most tolerability outcomes, whereas ubrogepant 50 mg ranked the best for these endpoints. It should be noted, however, that rankings do not measure the actual treatment effects and are only intended as an addition to the main outcomes reported above.
Treatment ranking based on P scores similar to SUCRA (surface under the cumulative ranking curve), where the ranking is presented as integer and the P score in brackets as percentages with 100% as the best possible and 0% the worst.

Discussion
In this review we evaluated the efficacy and safety of three novel drugs for the acute treatment of migraine attacks, namely lasmiditan, rimegepant and ubrogepant, in a network meta-analysis comparing recent phase 3 randomized controlled trials.
Our primary efficacy measures reflect the current IHS guidelines and thus considered as co-primary endpoints the percentage of participants who became pain free at two hours after treatment and the absence of the most bothersome migraine-associated symptom at two hours. All interventions in the included studies showed superiority to placebo for both endpoints. While lasmiditan at the higher doses (100 and 200 mg) was superior to all other treatments with respect to two-hour pain freedom, freedom from MBS was equally achieved with rimegepant and the commercially available doses of ubrogepant (50 and 100 mg).
Relevant secondary efficacy endpoints included freedom from migraine symptoms, which were photophobia, phonophobia and nausea. Lasmiditan 200 mg showed superiority in freedom from photophobia, however this was almost equally achieved with the 100 mg dose of the same drug, as well as rimegepant and ubrogepant 100 mg. In the freedom from phonophobia outcome rimegepant was equal to lasmiditan at high doses, whereas ubrogepant was not as effective. Freedom from nausea, on the other hand, showed larger estimated effects for rimegepant and higher doses of ubrogepant compared to placebo than for all lasmiditan doses, most likely because of the high incidence of nausea as a side effect associated with lasmiditan. Rimegepant and lasmiditan 200 mg were superior to other interventions for sustained pain freedom at 24 hours, whereas pain relief at two hours had the highest odds to be achieved compared to placebo with the higher doses of lasmiditan and with rimegepant. These efficacy results are similar to those of a recent network meta-analysis comparing lasmiditan with rimegepant and ubrogepant (30).
With regards to side effects, all doses of lasmiditan showed overall larger odds of developing treatment emergent adverse events compared to placebo, in particular dizziness, nausea and somnolence. This was confirmed in the extended safety findings publication for CENTURION (31) and in post-hoc analyses for SAMURAI and SPARTAN (32). Central nervous system side effects are a well-known issue with lasmiditan and believed to be due to its penetration of the blood-brain barrier (33) as well as possible localization of the 5-HT1F receptor in the cerebellar cortex (34). In fact, as lasmiditan has been found to impact significantly the ability to drive (35), a recommendation is made in the product characteristics to avoid driving or operating machinery for at least eight hours after each dose.
On the contrary, both formulations of rimegepant 75 mg and ubrogepant 50 mg showed a good ratio of efficacy vs side effects. A recent network meta-analysis performed on the same drugs including a total of five studies showed very similar results (36).
Limitations of this meta-analysis are that it was restricted to phase 3 randomized controlled trials and thus did not include efficacy and safety data from long-term open label or phase 2 studies. Further, study populations tended to be very homogeneous, as most studies excluded either non-responders, patients with medication overuse or patients with chronic migraine, limiting their generalizability. Some of these aspects are investigated with more detail in other papers within this Special Collection (37,38). Moreover, placebo response rate variations for two-hour pain free outcomes vary by nearly two-fold, which offers caution in terms of consistency of effect as a key assumption in network meta-analysis (39). The assessments of bias involve judgement (17) that, as constructed, have very poor qualities from an arithmetic viewpoint. Whether heterogeneity in the population effect is limited sufficiently to interpret the findings should be factored into any interpretation of the conclusions.
In fact, as the nature of a meta-analysis is that of indirect comparisons, our results should be regarded with caution as they cannot substitute direct comparative studies. These are particularly needed and should be directed to populations typically excluded from the phase 3 studies, such as patients in whom triptans are not effective or contraindicated because of cardiovascular diseases, who have the greatest unmet clinical needs.
The strength of this study is that it includes results from network meta-analysis as well as pairwise comparisons, which essentially allowed confirmation of our main findings. As a further validation of these analyses, we also used rankings, which provide the reader with a more generalized assessment of each drug. Further, all studies included in the meta-analysis were of high quality and did not present any bias risks, strengthening our results. Finally, by focusing equally on efficacy and safety/tolerability outcomes, our study allows for a more comprehensive evaluation of these treatments in comparison to existing literature, in an attempt that is similar to what has been done for triptans (40).
In the future, adequate head-to-head clinical trials that compare these different novel drugs in a direct manner would provide a further confirmation of these important findings. Whether these could represent value-for-money, or indeed what endpoints might be chosen remain issues to be considered.
Conclusions
This systematic review and meta-analysis allowed to uncover important aspects of the safety and efficacy profiles of novel treatments for migraine attacks, in the absence of direct comparative studies. In particular, lasmiditan, the 5-HT1F receptors agonist, was found to be more effective than placebo and, in higher doses, to both rimegepant and ubrogepant, for two-hour pain freedom. However, the increased link to central nervous system-related adverse events might limit its use in clinical practice. Rimegepant 75 mg and ubrogepant 50 and 100 mg present good efficacy and a favorable tolerability profile, albeit lower odds of achieving complete pain freedom. This profile might make these drugs the preferred choice for some patients, particularly for those who seek sustained relief from pain and nausea. These results should be nonetheless taken with caution, mostly due to heterogeneity and limited representativeness of the study populations, as well as the intrinsic limitation of indirect comparisons.
Article Highlights
This systematic review and meta-analysis allowed to determine a network of seven phase 3 RCTs (SAMURAI, SPARTAN, CENTURION, Study 302, Study 303, ACHIEVE I and II) involving n = 12,859 patients. The novel migraine drugs lasmiditan, rimegepant and ubrogepant are effective for the management of acute attacks and superior to placebo, as shown by high-level of evidence. Lasmiditan at high doses was the most effective treatment, although linked to significantly higher odds of adverse events such as dizziness, nausea and somnolence. Rimegepant and ubrogepant have higher tolerability rates and show similar levels of efficacy to lasmiditan in outcomes such as freedom from most bothersome symptom and freedom from photophobia.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024231151419 - Supplemental material for Efficacy, safety and indirect comparisons of lasmiditan, rimegepant, and ubrogepant for the acute treatment of migraine: A systematic review and network meta-analysis of the literature
Supplemental material, sj-pdf-1-cep-10.1177_03331024231151419 for Efficacy, safety and indirect comparisons of lasmiditan, rimegepant, and ubrogepant for the acute treatment of migraine: A systematic review and network meta-analysis of the literature by Francesca Puledda, Samaira Younis, Eva-Maria Huessler, Faraidoon Haghdoost, Marco Lisicki, Peter J Goadsby and Cristina Tassorelli in Cephalalgia
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Authors FP, SY, ML, FH, EMH declare no competing interest for this study.
Author PJG reports grants and personal fees from Amgen and Eli-Lilly and Company, and personal fees from Alder Biopharmaceuticals, Allergan, Autonomic Technologies Inc., Biohaven Pharmaceuticals Inc., Dr Reddy's Laboratories, Electrocore LLC, eNeura, Novartis, Teva Pharmaceuticals, and Trigemina Inc., and personal fees from MedicoLegal work, Massachusetts Medical Society, Up-to-Date, Oxford University Press, and Wolters Kluwer; and a patent Magnetic stimulation for headache assigned to eNeura without fee.
Author CT reports personal fees participating in advisor boards or lecturing at symposia from Allergan/Abbvie, Eli-Lilly and Company, Lundbeck, Novartis, and Teva Pharmaceuticals; institutional fees from clinical trials for Allergan/Abbvie, Eli-Lilly and Company, Lundbeck, Novartis, and Teva Pharmaceuticals.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.