
Abstract
Background
Epigenetic mechanisms, including DNA methylation, microRNAs and histone modifications, may modulate the genetic expression in migraine and its interaction with internal and external factors, such as lifestyle and environmental changes.
Objective
To summarize, contextualize and critically analyze the published literature on the current state of epigenetic mechanisms in migraine in a narrative review.
Findings
The studies published to date have used different approaches and methodologies to determine the role of epigenetic mechanisms in migraine. Epigenetic changes seem to be involved in migraine and are increasing our knowledge of the disease.
Conclusions
Changes in DNA methylation, microRNA expression and histone modifications could be utilized as biomarkers that would be highly valuable for patient stratification, molecular diagnosis, and precision medicine in migraine.
Keywords
Introduction
The term epigenetics was coined in 1942 by the developmental biologist Conrad H Waddington as bridging the gap between changes in phenotype with changes that do not incur in the genotype, to explain some aspects of development for which there was no understanding at that time (1). The knowledge around this term has greatly evolved and it is currently known that epigenetic mechanisms modify the inheritance of gene expression patterns without altering the underlying deoxyribonucleic acid (DNA) sequence through different reversible mechanisms (2), including DNA methylation, non-coding ribonucleic acid (RNA) regulation (such as microRNAs), and histone post-transcriptional modifications (chromatin remodeling) (3) (Figure 1).

Epigenetic mechanisms include histone post-transcriptional modifications (chromatin remodeling), DNA methylation and microRNAs (miRNAs) regulation.
DNA methylation
DNA methylation was the first epigenetic modification described in humans, hence it is the most well-studied epigenetic mechanism (4). This post-replication mark occurs when a DNA methyltransferase (DNMT) transfers a methyl group to the 5′ position of a cytosine, forming, a 5-methylcytosine (5-mC). Most of the DNA methylation occurs within repeated CpG dinucleotide regions, called CpG islands, which are usually located in the promoter region of a gene. This modification leads to gene expression silencing due to inhibition of the required transcription factor(s) binding to the promoter (5). These modifications can also be found in intragenic regions, although their exact role has not been established yet (6). The DNA methylation print is stable and can be inherited during development and cell differentiation (7), although changes in global DNA methylation seem to occur naturally not only in aging (8) but also due to environmental and lifestyle factors (9).
DNA methylation has been found to play an important role in psychiatric and neurological disorders. It has been shown that patients with autism spectrum disorder (ASD) and Rett Syndrome have a specific DNA methylation pattern in the promoter of the Methyl-CpG-binding protein 2 (MECP2) gene that leads to the differential expression of genes related to synaptic activity and brain derived neurotrophic factor (10,11). Also, there is evidence in Alzheimer’s disease (AD) that DNA is significantly hypomethylated in the promoter regions of the β-amyloid (Aβ) peptide generation-related genes (APP, PS1 and BACE1) (12–14); and in Parkinson’s disease (PD), that there is a significant hypomethylation of the SNCA and PARK2 promoters (genes related to the biosynthesis of α-synuclein) (15).
Non-coding RNA
Among the epigenetic regulatory mechanisms, microRNAs (miRNA) are a subtype of non-coding RNA of ∼22 nucleotides long that act as key regulators of genetic expression by inhibiting transcription or promoting degradation of selected messenger RNAs (mRNAs), which is a way of controlling gene expression, doing so by binding to the 3′-untranslated region (UTR) of target mRNA. miRNAs are transcribed by the RNA polymerase II/III in the nucleus, forming the primary miRNA (pri-miRNA). Then, pri-miRNA is processed and exported to the cytoplasm by exportin 5 and, after subsequent processing by the RNase III enzyme Dicer, a small RNA duplex (mature miRNA) that directly binds to the ribosome/mRNA complex is released. A single miRNA sequence regulates multiple mRNA, exerting a pleiotropic modulation of cellular processes (16). Interestingly, it has been stated that miRNA could also help mediate the environmental induced changes in genetic expression (17). Current literature reports differentially expressed miRNA in all body fluids, including blood, saliva, urine and CSF, making miRNA an ideal candidate biomarker (18).
Their expression can reflect changes in the brain, including multiple central nervous system (CNS) processes and changes in neurotransmitters and hormones (19). Same as with DNA methylation modifications, differential miRNA expression profiles have been identified in mental disorders and neurological diseases. Altered expression of some miRNA in patients suffering from AD suggested that miRNA may have a crucial regulatory role on the mechanisms involved in its pathogenesis, including Aβ metabolism by modulating the activity of the β-secretase BACE1 and tau aggregation (20). In PD patients, multiple miRNAs were associated with α-synuclein-mediated neurotoxicity (21). There is also strong evidence that miRNA dysregulation is linked to a differential modulation of the process of stroke (22) and to the mechanisms of epileptogenesis (23), highlighting the relevance of the role of miRNAs in neurological pathogenesis.
Histone modifications and chromatin remodeling
As an epigenetic modification, post-transcriptional histone marks do not modify the sequence of DNA but can modify its expression regulating the transcriptional process. The main histone modifications associated with the regulation of chromatin structure and, therefore, transcriptional activity are acetylation, methylation, phosphorylation, and ubiquitination (24). Histone modifications are mainly done at the N-terminal tails of amino acids by a specific enzymatic machinery. Depending on the modification and the histone involved, the final effect might be activating or repressing gene expression (25).
A recent study has shown that abnormal histone modifications can affect neurodevelopmental processes and cause a series of neurological diseases (26). For instance, mutations of histone demethylase KDM5C are associated with ASD phenotype, as well as with aggressive behavior (27).
Lifespan migraine progression: From risks factors to epigenetic resilience
Migraine is a complex neurological disorder that strongly aggregates in families (28). This observation has been the rationale to explore underlying genetic factors. In the last decade, the fast advancement of genetic high-throughput technologies has allowed the use of large-scale genome-wide association studies (GWAS) to assess the genetics of migraine, the results of which have pointed towards describing migraine as a complex polygenic disorder. Despite the success of these studies, which have identified 123 risk loci for migraine which mainly codify for neuronal and vascular functions (29), the evidence describes migraine as a lifelong disorder that affects more women than men, which can increase in frequency and transform, through a chronification process, into chronic migraine (CM), where patients’ burden increases exponentially (30).
Although ineffective acute and preventive treatment, and the consequent insufficient pain relief, has been highlighted as one of the highest risk factors for disease chronification, other factors such as female sex, low educational status, stressful life events and the presence of comorbidities have also been associated to migraine transformation (31,32). Even though the mechanisms behind this transformation are not fully understood, it has been suggested that patients with CM might have lower sensory thresholds which among other factors lead to a higher susceptibility to triggering migraine attacks, which in turn, can also facilitate the recurrence of attacks as frequency increases (33). Conversely, for some CM patients, the disease state is reversible: a 26% of patients with CM remit to the episodic form within two years (34). Indeed, migraine frequency cycles, and we still do not understand why this happens and who is more susceptible (35). Altogether, genetic predisposition, accumulated risk factors, abnormal neurophysiological information processing, morphological brain changes and disease reversible processes point to altered neural plasticity mechanisms that can only be contributed to epigenetic pathways.
DNA methylation in migraine
Clinical studies
Four different studies have analyzed the potential role of DNA methylation in migraine patients, by using DNA from peripheral blood, to explore the existence of peripheral biomarkers associated with this epigenetic modification, a summary can be found in Table 1. Wan et al. (36) explored the potential implication of the DNA methylation of the calcitonin gene-related peptide (CGRP) receptor subunit RAMP1 in migraine. To achieve this, they analyzed the methylation levels in the promoter of RAMP1 in 26 migraine patients and 15 healthy controls. However, their analysis did not retrieve any significant differences between both groups, which could be partly due to the low sample size of the study. In a more recent paper, Gerring et al. (37) performed an epigenome-wide association study by quantifying genome-wide patterns of DNA methylation in 67 migraine patients and 67 healthy controls by using the Illumina HumanMethylation450 BeadChip array. The analyses conducted between migraine and methylation probe expression did not show any significant association after correction for multiple testing. Hence, authors used a 1kb sliding window approach to combine adjacent migraine-methylation association P values, which allowed them to identify 62 independent differentially methylated regions that were associated with the disease. Further analyses of these regions indicated that they were enriched in regulatory elements of the genome, including CpG islands and shores and transcription start sites, and that they were in close proximity to genes involved in solute transportation or hemostasis.
Summary of the epigenetic studies performed in migraine.
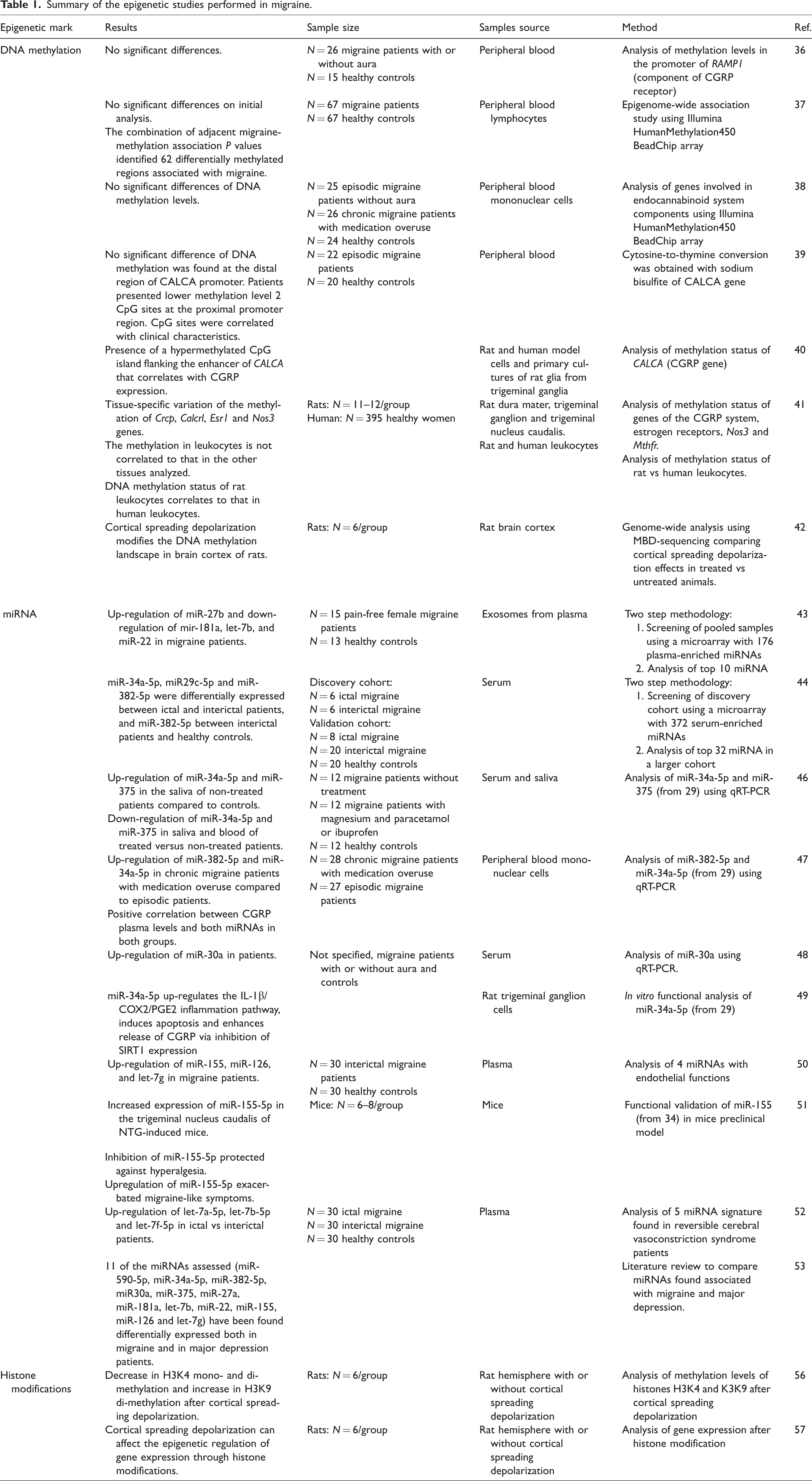
Greco et al. (38) used the same technology approach, the Illumina HumanMethylation450 BeadChip array, to analyze a cohort of 25 episodic migraine patients without aura, 26 chronic migraine patients with medication overuse and 24 healthy controls. In this study, the authors evaluated changes in the DNA methylation status of those genes involved in endocannabinoid system components, together with an analysis of gene and protein expression of them, with the aim to identify a specific expression pattern of the endocannabinoid system at the peripheral level in migraine patients. Interestingly, they found a higher gene and protein expression of the cannabinoid receptors 1 and 2 (CB1 and CB2) and a lower gene expression of the fatty acid amide hydrolase (FAAH) and the N-arachidonoyl phosphatidylethanolamine phospholipase D (NAPE) genes in episodic and chronic migraine patients when compared to controls. However, they did not find any significant differences at the DNA methylation levels of all the evaluated genes, which suggested that the changes in gene expression could be related to other epigenetic mechanisms.
Finally, in the last study of DNA methylation in migraine, Rubino et al. (39) evaluated the DNA methylation status of the promoter region of CALCA, the gene encoding CGRP, by using bisulfide sequencing, in 22 episodic migraine patients and 20 healthy controls. Authors found lower methylation levels in 2 CpG sites at the proximal promoter region in patients, localized within the binding site of the transcription factor CREB, suggesting an increased transcription of the gene. Moreover, they found a correlation between the levels of methylation of CALCA and several clinical features of migraine, including age at onset, presence of nausea/vomiting, depression, and anxiety.
In vitro, in vivo and preclinical studies
Other studies, summarized in Table 1, have focused on analyzing the DNA methylation of genes related to migraine either in vitro or in vivo in tissues that are related to migraine pathophysiology, to overcome the limitation of the tissue-specificity of epigenetic modifications.
In a study led by Park et al. (40), the DNA methylation status of CALCA was measured in vitro in rat and human model cells and in primary cultures of rat glia from the trigeminal ganglia. They were able to show the presence of a hypermethylated CpG island that flanked the enhancer of CALCA in the three cell lines and that was correlated with a lack of expression of the gene. Subsequent treatment with the DNA methylation inhibitor 5-aza-2′-deoxycytidine induced an increased expression of CALCA mRNA in glial cultures, indicating that changes in the DNA methylation of the enhancer of the gene are sufficient to modulate its expression. Future studies replicating these results in vivo could be convenient to demonstrate the possibility to down-regulate the expression of CGRP using epigenetic mechanisms as an alternative treatment for migraine patients.
Another study analyzed the methylation status of migraine-related genes in migraine-related tissues (including dura mater, trigeminal ganglion and trigeminal nucleus caudalis) of the rat and compared them with their methylation status in leukocytes and aorta, along with an analysis of the effect of 17β-estradiol on the methylation of these genes (41). The study was focused on genes of the CGRP-ergic system, including Calca, Ramp1, Crcp, Calcrl, Usf2; estrogen receptors, including Esr1 and Gper; and the nitric oxide synthase 3 (Nos3) and Mthfr genes. Their results showed that the methylation of Crcp, Calcrl, Esr1 and Nos3 genes highly varies according to the tissue analyzed, that treatment with 17β-estradiol does not modify the methylation status of the studied genes and that the methylation in leukocytes is not correlated to that in other tissues, highlighting one of the challenges of studying epigenetic marks in migraine.
Of note, further analyses of this study showed that the DNA methylation status of rat leukocytes was similar to the one seen in human leukocytes in the studied genes, suggesting the appropriateness of using the rat as a model to study human DNA methylation when there is a difficulty in obtaining the target tissue. On this line, a different study evaluated whether cortical spreading depolarizations (CSDs), the plausible origin of migraine aura, could be modulating DNA methylation at a genomic level in cortical tissue of rats and whether this would be modified by chronic treatment with topiramate or valproate (42). Their analyses showed a differential methylation of genes after inducing CSD that was further modified by both treatments. Although this study has some limitations, such as that rats underwent multiple CSDs, the results might suggest that migraine aura could alter the epigenetic landscape of the cortex through changes in DNA methylation that might, in turn, increase the susceptibility to suffer subsequent episodes of migraine aura.
microRNA expression changes in migraine
Several pilot studies, summarized in Table 1, have described miRNAs as potential peripheral biomarkers of migraine. Of these, only two studies have followed a hypothesis-free methodology to discover miRNAs associated to migraine (43,44). In the first of them, Tafuri et al. (43) used a two-step methodology to uncover differences in miRNA expression of exosomes (exo-miRNAs) isolated from plasma between pain-free female migraine patients (N = 15) and healthy controls (N = 13). Exosomes are endosome-derived extracellular nanovesicles that are released into biological fluids (plasma, serum and cerebrospinal fluid) and act as carriers of various molecules, including miRNAs. Exo-miRNAs are highly stable, making them a promising source of biomarkers (45). Tafuri et al. pooled the samples in each group and performed an initial screening using a microarray that covered 176 miRNAs that are known to be highly enriched in plasma. Subsequently, they further assessed by qRT-PCR the 10 miRNAs that showed a differential expression between both groups, including let-7b, miR-22, miR-26a, miR-26b, miR-27b, miR-29b, miR-30b, miR-30e, miR-181a, and miR-221. This led the authors to find four miRNAs that were significantly differentially expressed in their cohort of female migraine patients: miR-27b was up-regulated, whereas mir-181a, let-7b, and miR-22 were down-regulated.
In the second study, Andersen et al. (44) used a two-step analysis to discover serum miRNAs differentially expressed at different phases of migraine attacks: ictally (2 h after the onset of pain) and interictally (at least after five days being pain-free) and compared them to healthy controls. They used a microarray containing 372 serum miRNAs for their first screening, which included six ictal and six interictal patients. This analysis retrieved 32 miRNAs that were differentially expressed during migraine attacks. From this list, authors selected four miRNAS to be validated in a cohort of patients with migraine (N = 8 ictal, N = 20 interictal) and in healthy controls (N = 20), a cohort that also included the initial screening cohort. This analysis showed that miR-34a-5p, miR29c-5p and miR-382-5p were differentially expressed between ictal and interictal patients, and miR-382-5p between interictal patients and healthy controls. Of note, authors did not perform any correction for multiple comparisons and, as stated above, the sample sizes used were low. These limitations, together with the facts that the study led by Tafuri et al. (43) analyzed miRNAs from exosomes and used a different microarray for miRNA screening, may be the reasons that could explain the divergence of the outcomes of both studies.
More recent studies have attempted to validate specific miRNAs selected from the Andersen et al. (44) study in their own cohorts. Gallelli et al. (46) evaluated the expression of miR-34a-5p and miR-375 using qRT-PCR in serum and saliva of migraine without aura patients, including non-treated patients (N = 12) and chronically-treated patients with magnesium and paracetamol or ibuprofen (N = 12); and compared them with healthy controls (N = 12). Their results showed an up-regulation of miR-34a-5p and miR-375 in the saliva of non-treated patients compared to controls and a down-regulation of miR-34a-5p and miR-375 in saliva and blood of treated versus non-treated patients. Although these results should be interpreted with caution especially due to the low sample size used, the results suggest that both miRNAs are expressed in blood and saliva and that they could be potentially used as biomarkers of disease. In a different study, Greco et al. (47) found higher serum levels of miR-382-5p and miR-34a-5p in chronic migraine patients with medication overuse (N = 28) when compared to episodic migraine patients (N = 27). Interestingly, they also showed a positive correlation of CGRP plasma levels with both miRNAs in the overall population, indicating a possible regulatory action of the miRNAs. Zhai and Zhu (48) allegedly selected miR-30a from the Andersen et al. study (44), although this specific miRNA was not found differentially expressed in their screening nor in their validation cohort, so it is unclear how the authors selected this specific miRNA. Interestingly, in this study they found a differential expression of serum miR-30a in an unspecified number of migraine patients with and without aura. Zhang et al. (49), instead, used an in vitro approach to determine the functional properties of miR-34a-5p. Specifically, they used isolated primary rat trigeminal ganglion neurons to show that miR-34a-5p up-regulates the IL-1β/COX2/PGE2 inflammation pathway, induces apoptosis and enhances release of CGRP via inhibition of SIRT1 expression. These results indicate a potential mechanism of action of miR-34a-5p that could explain some aspects of migraine.
A different study used a candidate approach to determine the potential effects of their pre-selected miRNAs in migraine. In this exploratory study, Cheng et al. (50) evaluated whether four plasma miRNAs with endothelial functions (miR-155, miR-126, miR-21, and let-7g) were altered in interictal migraine patients without overt vascular risk factors (N = 30) when compared to healthy controls (N = 30). Interestingly, their results showed significant up-regulated expression of miR-155, miR-126, and let-7g in migraine patients when compared to controls. However, these results should be validated in larger and independent cohorts of patients. Of note, one of these miRNAs, miR-155, was functionally studied in the nitroglycerin-induced chronic migraine mouse model in a more recent work (51). Once the model had been established and mice showed an increased level of hyperalgesia when compared to controls, authors were able to show an increased expression of miR-155-5p in the trigeminal nucleus caudalis (TNC). The administration of a miR-155-5p antagomir (an inhibitor of the miRNA) protected against the development of the nitroglycerin-induced hyperalgesia and, on the same line, the up-regulation of miR-155-5p led to exacerbated migraine-like symptoms, suggesting the implication of this miRNA in the development of central sensitization in the nitroglycerin-induced mouse model of migraine.
In another two studies, authors studied the similarity of the aberrant expression of miRNAs in migraine compared to other disorders, including reversible cerebral vasoconstriction syndrome and major depression, to understand the comorbidities of migraine. The former was assessed by Chen et al. (52) that used a cohort of 60 episodic migraine patients, that included 30 ictal and 30 interictal patients, and 30 healthy controls to examine the specificity of a 5 miRNA signature found in reversible cerebral vasoconstriction syndrome patients. Their analysis showed that let-7a-5p, let-7b-5p and let-7f-5p were up-regulated in ictal migraine patients when compared to interictal patients and controls, suggesting an association of these miRNAs with acute headache or potentially indicating the presence of shared mechanisms between reversible cerebral vasoconstriction syndrome and migraine. The link between migraine and major depressive disorder was assessed by Chen et al. (53) using a different approach. In this case, they reviewed the literature and compared the miRNAs that had been previously found associated to migraine with those associated to major depression. Interestingly, they found that 11 of the miRNAs assessed (including miR-590-5p, miR-34a-5p, miR-382-5p, miR30a, miR-375, miR-27a, let-7b, miR-22, miR-155, miR-126 and let-7g) had been found differentially expressed both in migraine and in major depression patients.
Histone modifications
Currently, there are no published studies that have focused on analyzing histone modifications in migraine patients. Although it is interesting to note that valproate, a preventive treatment for migraine (54), is a well-known histone deacetylase inhibitor (55). Hence, future studies could investigate whether the beneficial effects of this treatment on migraine patients could be related to the modification of histones.
Despite the lack of clinical studies analyzing whether histone modifications have a role or not in migraine, two different preclinical studies from the same group have used rat models to analyze the potential effects of CSD on histone methylation. In the initial study Passaro et al. (56) compared the methylation levels of the histones H3K4 and H3K9 between the hemispheres of rats where CSDs were induced and in the contralateral ones. The analysis retrieved significant decreases in H3K4 mono- and di-methylation and increased H3K9 di-methylation. These changes were further assessed in a later study where the authors confirmed that CSD can affect the epigenetic regulation of gene expression through histone modifications (57), a finding that will be hard to replicate in humans due to the difficulty in obtaining cortical tissue after experiencing a migraine aura.
The role of epigenetics in personalized medicine for migraine: Relevance and challenges
Emerging evidence indicates that epigenetic mechanisms are crucial for the correct development and maintenance of brain functions, and that defects in these mechanisms can alter disease susceptibility, contribute to their pathophysiology and determine the response to specific treatments (58). Moreover, epigenetic biomarkers can be used as diagnostic tools for clinical practice (59). They are a simple method to associate molecular markers with contributions of lifestyle and environment in disease, adding an extra layer of information to the genetic markers. Moreover, epigenetic changes, especially DNA methylation, are stable in fluids and tissue that are routinely used in clinical settings facilitating the methodologies used to study them; they can be detected at early stages of disease; and they have been shown to be robust, sensitive and measurable across individuals and populations (60).
However, there are several challenges of using epigenetic biomarkers that need to be considered. Although epigenetic modifications confer a reversible adaptation of the genome to the internal and external environment, they have a strong cell- and tissue-type dependence; hence, the epigenetic status between different tissues, for example brain and blood, has been found to differ in most cases, although there are some studies that indicate the opposite (61), or that even indicate a better correlation between brain tissue and saliva (62). Another important challenge is that epigenetic mechanisms also happen during normal development and differentiation (63), and this variation should be accounted for when comparing samples from different sources.
The above-mentioned points highlight the relevance and challenges of studying epigenetic mechanisms in migraine and suggest that their abnormalities could explain some of the differences reported in migraine patients, including the differences in their frequency of attacks, their susceptibility to migraine chronification and their response to anti-migraine drugs, among others. Indeed, epigenetic abnormalities have been frequently found in neurological disorders, including epilepsy (64), Parkinson disease (65), Alzheimer disease (66), and amyotrophic lateral sclerosis (67); in neuropathic pain (68), and in psychiatric disorders, such as major depressive disorder (69); although many potential biomarkers have been proposed in neurological diseases without being translated into clinical settings, probably because of the difference of the epigenetic status between target/study tissue and the lack of consideration of other factors that modify the epigenetic signature, such as lifestyle (diet, exposure to chemicals, smoking) and internal factors (disease subtypes, symptoms, genetic markers, treatments, age) (70). All evidence gathered in this narrative review seems to support a role of epigenetic processes involved in mechanisms of brain plasticity and migraine-specific processes.
Discussion
There is supporting evidence that epigenetic mechanisms, including, but not limited to, DNA methylation, miRNAs and histone modifications, have a role in migraine. The studies performed to date, which have been reviewed here, have analyzed these mechanisms to evaluate their implications in different aspects of the disease, comparing episodic vs chronic migraine, treated vs untreated patients; and also, looking into migraine with other diseases, and their use in preclinical models. Although most of them point towards supporting a role of epigenetic mechanisms in migraine, there are limitations that complicate the reproducibility of their results, and as such, these studies should be considered as exploratory.
Future studies including bigger and more homogeneous cohorts will certainly increase our understanding of the role that epigenetic mechanisms may play in migraine susceptibility, and they can also benefit from the recent advancement, and lowered cost, of the methodologies to study epigenetics at a genomic level. However, efforts should be taken in future studies to standardize protocols, including sample size, type of patients and controls included with an appropriate balance of gender, and homogenous DNA, RNA and protein extraction protocols, to obtain more reliable outcomes that might improve our knowledge of the molecular processes that underlie migraine.
Finally, the use of blood or saliva to study epigenetic mechanisms involved in migraine may allow for the identification of biomarkers that would be highly valuable for patient stratification, diagnosis, and precision medicine (71). However, due to the uncertainty of the epigenetic concordance between brain tissue and blood or saliva (70), it would be interesting to compare the results in different tissues and to functionally study the biomarkers found to better understand their biological significance.
Key findings
Epigenetic changes measured with microRNAs, DNA methylation or histone modifications have been found in animal models and patients with migraine. Epigenetic changes in migraine can help diagnose and stratify patients. Epigenetic changes in migraine could be considered as a biomarker of disease; this field of study deserves further research, as it might lead to practical implications on management and treatment of migraine.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MV-P was supported by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie IF grant agreement No. 101023175.