
Abstract
Background
Atogepant is a United States Food and Drug Administration-approved oral calcitonin gene-related peptide receptor antagonist for the preventive treatment of episodic migraine. The study objective was to evaluate the long-term safety and tolerability of atogepant in participants who completed the phase 3 ADVANCE trial (NCT03777059).
Methods
This 40-week, open-label extension trial (NCT03939312) monitored safety in participants receiving oral atogepant 60 mg once daily, followed by a four-week safety follow-up period.
Results
Of the 685 participants taking at least one dose of atogepant, the treatment period was completed by 74.6% of participants with a mean (standard deviation) treatment duration of 233.6 (89.3) days. Treatment-emergent adverse events occurred in 62.5% of participants, with upper respiratory tract infection (5.5%), urinary tract infection (5.3%), nasopharyngitis (4.8%), sinusitis (3.6%), constipation (3.4%), and nausea (3.4%) occurring at ≥3%. Serious adverse events were observed in 3.4% of participants (none were treatment-related), and there were no deaths. Adverse events leading to discontinuation occurring at >0.1% were nausea (0.4%) and abdominal pain, vomiting, weight decrease, dizziness, and migraine (0.3% each).
Conclusion
These results are consistent with atogepant’s known safety profile and support long-term use of atogepant 60 mg once daily dosing as safe and well tolerated.
ClinicalTrials.gov Registration Number: NCT03939312
Introduction
According to the 2019 Global Burden of Disease study data, the prevalence of migraine is approximately 1 billion cases worldwide, with 42 million years lived with disability (YLDs) globally (1). Migraine is the second leading cause of YLDs worldwide (2) and the leading cause in adults under 50 (3).
In clinical practice, patients using older migraine preventive therapies, such as antidepressants, anticonvulsants, or beta-blockers, have poor treatment adherence rates, with reasons for discontinuation most often cited as a lack of efficacy or due to side effects of the medications (4,5). Furthermore, only 3 to 13% of migraine patients use preventive treatments (6). An American Headache Society 2019 consensus statement suggests that integrating new migraine treatments into current clinical practice would benefit approximately 40% of patients with episodic migraine (6).
Calcitonin gene-related peptide (CGRP) antagonists have been developed for the acute or preventive treatment of migraine and provide positive efficacy in migraine prevention with low adverse event rates. However, the lack of long-term data may hamper the assessment of benefit-risk profiles before treatment initiation (7). In addition, because patients with migraine may need multiple years of treatment, preventive treatments for migraine should demonstrate favorable long-term benefit-risk profiles (8). A 52-week study with atogepant helps to shed light on long-term safety and tolerability of atogepant (9), but additional long-term safety analyses of CGRP inhibitors are needed.
Atogepant is an oral small-molecule CGRP receptor antagonist approved by the US Food and Drug Administration (FDA) in 2021 for the preventive treatment of episodic migraine in adults (10). Two 12-week, double-blinded, placebo-controlled, randomized clinical trials (phase 2b/3 [NCT02848326] and phase 3 ADVANCE [NCT03777059] [11,12]) demonstrate that atogepant is effective and well tolerated in the preventive treatment of episodic migraine. Results from the 12-week pivotal trials (11,12) demonstrate that oral atogepant 60 mg once daily treatment administered for the prevention of episodic migraine is safe and well tolerated. The primary objective of this long-term open-label extension trial was to evaluate the safety and tolerability of oral atogepant 60 mg treatment administered once daily for 40 weeks after participant completion of the phase 3 ADVANCE trial for the preventive treatment of episodic migraine.
Methods
This was a multicenter, open-label, long-term safety extension (309-OLEX) of the pivotal ADVANCE phase 3 trial (Figure 1), which screened 695 participants from 116 sites across the United States (NCT03939312). The first visit of any participant was 6 May 2019, and the last visit of any participant was completed on 31 March 2021. The protocol and all amendments were approved by the institutional review boards (IRB) for each trial center (online Supplemental Table S1); all participants provided written informed consent. Investigators could not implement changes to the protocol without prior review and approval from the local IRB, except where necessary to reduce or eliminate risks to trial participants. All trial conduct was in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. Data were analyzed by the trial sponsor, AbbVie Inc. (formerly Allergan). All authors had full access to the trial data.

Trial Design.
Trial participants
Adult participants with episodic migraine who completed the lead-in trial were eligible to participate in this long-term safety extension trial (Figure 1), with most 309-OLEX trial participants having little to no gap in atogepant treatment (online Supplemental Table S2). Non-compliance with ADVANCE trial protocol-required procedures or ADVANCE trial adverse events that indicated an unacceptable safety risk affected participant eligibility for the 309-OLEX trial.
Participants were excluded from the 309-OLEX trial if they presented with clinically significant cardiovascular, endocrine, gastrointestinal, hematologic, hepatic, neurologic, pulmonary, or renal disease or had a condition that could significantly interfere with trial participation, confound trial results, or put the participant at significant safety risk, as determined by the investigator. Participants were also excluded if they exhibited clinically significant electrocardiogram (ECG) abnormalities, hypertension as defined by a sitting systolic blood pressure >160 mmHg or a sitting diastolic blood pressure >100 mmHg, or if female participants had a positive urine pregnancy test. Furthermore, women of child-bearing age who were pregnant, lactating, or were planning to become pregnant during the course of the trial were excluded. Participants were excluded based on their answers to the Columbia Suicide Severity Rating Scale (C-SSRS) questions if they exhibited a significant risk of self-harm or harm to others in the opinion of the investigator. Participants who did not adhere to concomitant medicine and dietary restrictions were also excluded (online Supplemental Table S3). CGRP-blocking monoclonal antibodies and botulinum toxin were prohibited for six months prior to Visit 1 and throughout the trial (online Supplemental Table S3). Allowable medication alternatives are listed in online Supplemental Table S4. No other gepants were taken during the open-label treatment period.
Trial design, outcomes, and assessments
The trial consisted of a 40-week open-label treatment period, followed by a four-week non-treatment safety follow-up period. The primary objective was to evaluate the safety and tolerability of treatment with oral atogepant 60 mg tablets, administered once daily for the preventive treatment of migraine in participants with episodic migraine. Participants were instructed to take the atogepant tablets at approximately the same time each day. Trial visits during the open-label treatment period were scheduled every four weeks after the first visit, with an end-of-study visit four weeks after the last dose of atogepant (Figure 1). All participants were allocated doses of 60 mg atogepant, with no control intervention and no blinding. An internet web-based recruiting system was responsible for managing atogepant inventory, dispensing atogepant, and providing confirmation notifications of each transaction. Electronic case report forms collected all office visit data via an electronic data capture system.
All safety analyses were performed on the safety population (participants who received at least one dose of atogepant during the 309-OLEX trial). Safety outcomes assessed included adverse events (AEs), clinical laboratory evaluations, the C-SSRS, vital sign measurements, physical examinations, and ECG parameters. All AEs were recorded at each visit from the time of informed consent to the final visit. Adverse event duration, severity, seriousness, causal relationship to atogepant, and treatment measures or actions taken to resolve the AE were documented. For clinical laboratory evaluations, non-fasting blood tests, urinalysis, and drug screenings, a centralized clinical laboratory was used to analyze all blood and urine samples (serology and urine drug screen only occurred at Visit 1).
Suicidal ideation and behavior severity was assessed by a clinician using the C-SSRS at each visit, which contains a 5-item scale for each ideation and behavior. Vital sign measurements performed at each visit included the parameters of weight, temperature, sitting and standing pulse rates, and blood pressure while sitting and standing. On visits 1, 4, 7, and 11, a 12-lead ECG was performed, transmitted to the centralized clinical laboratory to be read by a cardiologist, and the ECG's clinical significance was recorded in the participant's case report form. Lastly, the baseline value (defined during the lead-in ADVANCE trial) was used as the comparator for the long-term 309-OLEX trial for each clinical laboratory evaluation, C-SSRS, vital sign measurement, and ECG parameter assessed.
Statistical analysis
No separate sample size calculation was performed for the trial extension, as all eligible participants from the lead-in trial were enrolled. Approximately 750 participants were expected to enroll in this extension trial based on the lead-in trial anticipated completion rate. All safety analyses were performed in the safety population consisting of participants who received at least one dose of atogepant in this study. The number and percentage of participants were used to summarize all categorical values, while descriptive statistics were used to summarize continuous variables. No efficacy analysis was conducted, as efficacy measures were not collected for this study.
Safety monitoring
Interim data analyses were performed to monitor safety during the 309-OLEX trial. An independent Data Safety Monitoring Board was established to identify any emergent unexpected, deleterious, or clinically significant safety issues, trends, or adverse events. An external Clinical Adjudication Committee was established to standardize surveillance and monitoring, and to determine if post-treatment elevations in hepatic-related laboratory values (such as alanine or aspartate aminotransferase [ALT or AST, respectively]) were related to atogepant treatment. Recommendations to the site investigator or the trial sponsor for trial modification or early termination could occur at any time based on recommendations from these groups; or, the site investigator or the trial sponsor could halt trial participation at any time.
Post-initiation changes
An amendment to the trial protocol was created after initiation to mitigate risks due to COVID-19. Trial staff were allowed to conduct remote visits as needed for up to eight weeks; participants unable to attend an in-person visit after eight weeks were discontinued. The final trial visit was conducted remotely for all participants. Adjustments to sponsor monitoring and oversight were instituted per FDA guidance on conducting trials during the COVID-19 pandemic (13). This protocol amendment included one statistical analysis plan amendment, which provided a remote assessment schedule and procedures for remote visits conducted during the COVID-19 pandemic.
Results
Participant disposition and exposure
From May 2019 to March 2021, 116 US sites screened 695 participants, with 685 participants entering the long-term open-label extension (309-OLEX) trial and taking at least one dose of atogepant (the Safety Population, Figure 2). Of the 10 participants excluded from entering the trial, most were excluded due to being lost to follow-up (Figure 2). Participant demographics and baseline characteristics in the Safety Population are listed in Table 1. Participants were 88.2% female, 84.4% white, with a mean age of 41.8 (standard deviation [SD], 12.3) years, a mean body mass index (BMI) of 30.58 (SD, 7.82) kg/m2, and a migraine diagnosis duration of >20 years (with or without aura). The open-label treatment period was completed by 74.6% of participants, with the safety follow-up period completed by 91.1% of the safety population. Completion was defined as those participants who attended the final scheduled visit. The two most common reasons for discontinuation were meeting withdrawal criteria after Visit 1 (6.3%, Labs and ECG) or withdrawal by participant (5.1%) in the open-label treatment period. The most common reasons for discontinuation during the safety follow-up period were participant withdrawal (1.8%) or lost to follow-up (0.6%, Figure 2). Protocol deviations were most often due to a participant taking a prohibited concomitant medication (15%) and participants remaining on trial intervention for longer than eight weeks without an onsite visit due to COVID-19 (6.7%).

Participant Disposition.
Participant demographics and baseline characteristics (safety population).

309-OLEX, open-label extension trial; BMI, body mass index; N, number of participants in the safety population; n, number of participants in the specific category; SD, standard deviation.
aThe last non-missing safety assessment before the first dose of treatment in the lead-in ADVANCE trial was used as the baseline in the current 309-OLEX trial.
bParticipants who reported multiple races are only included in the “More than one race” category.
The mean treatment duration of once-daily oral atogepant 60 mg was 233.6 (SD, 89.3) days, ranging from one to 319 days, with a median of 280 days (upper quartile of 281 days and a lower quartile of 257 days). Taken together, this translates to 438.1 participant-years of atogepant exposure, with 78.5% of participants exposed for ≥180 days (at least six months) and 74.3% of participants exposed for ≥270 days (at least nine months).
Treatment-emergent adverse events
The overall summary of treatment-emergent adverse events (TEAEs) in the safety population is shown in Table 2. Nearly 63% of participants reported TEAEs (28.6% mild, 30.2% moderate, and 3.6% severe) in this long-term 309-OLEX trial safety population. Table 3 lists all TEAEs that occurred in ≥2% of atogepant-treated participants, with the TEAEs occurring at ≥3% being upper respiratory tract infection (5.5%), urinary tract infection (5.3%), nasopharyngitis (4.8%), sinusitis (3.6%), constipation (3.4%), and nausea (3.4%). In the 428 participants who reported an adverse event, 8.8% of the safety population were determined to have experienced a treatment-related adverse event (per the investigator). The most frequent (>1%) treatment-related adverse events were constipation (n = 11, 1.6%), nausea (n = 8, 1.2%), and weight decrease (n = 8, 1.2%). TEAEs led to discontinuation in 3.2% of the safety population. Table 4 lists all TEAEs leading to discontinuation that occurred in >0.1% of participants (nausea, abdominal pain, vomiting, weight decrease, dizziness, and migraine). No deaths occurred during the long-term 309-OLEX trial.
Overall summary of treatment-emergent adverse events (safety population).
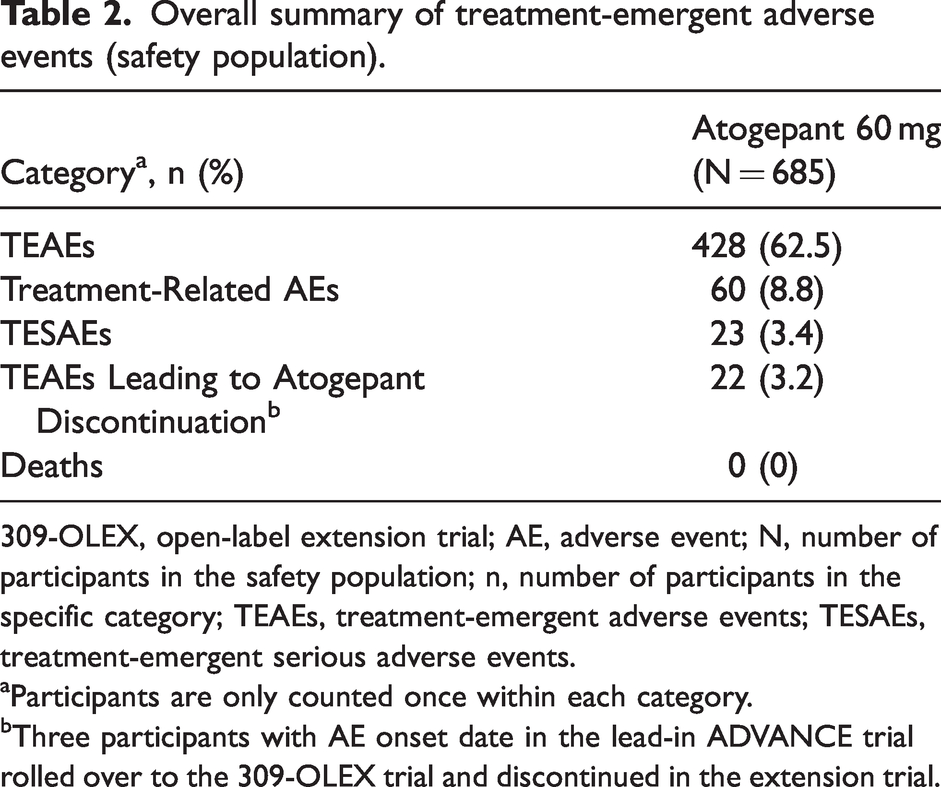
309-OLEX, open-label extension trial; AE, adverse event; N, number of participants in the safety population; n, number of participants in the specific category; TEAEs, treatment-emergent adverse events; TESAEs, treatment-emergent serious adverse events.
aParticipants are only counted once within each category.
bThree participants with AE onset date in the lead-in ADVANCE trial rolled over to the 309-OLEX trial and discontinued in the extension trial.
Treatment-emergent adverse events (≥2%) by preferred term (safety population).
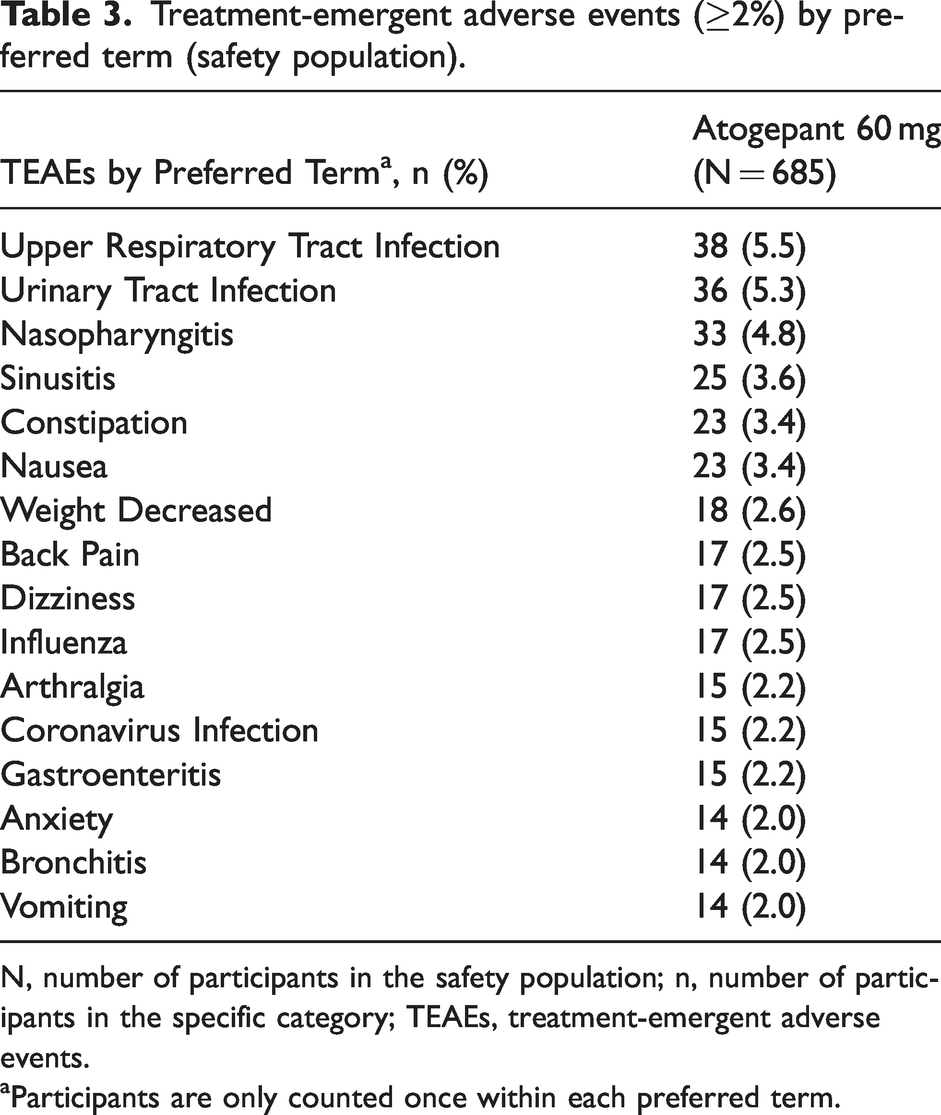
N, number of participants in the safety population; n, number of participants in the specific category; TEAEs, treatment-emergent adverse events.
aParticipants are only counted once within each preferred term.
TEAEs (>0.1%) leading to atogepant discontinuation by preferred term (safety population).

N, number of participants in the safety population; n, number of participants in the specific category; TEAEs, treatment-emergent adverse events.
aParticipants are only counted once within each preferred term.
Although 3.4% of trial participants experienced a treatment-emergent serious adverse event (TESAE), none were related to atogepant treatment by the investigators. The most frequent (>0.1%) TESAEs are listed in Table 5.
Serious TEAEs (>0.1%) by preferred term (safety population).

N, number of participants in the safety population; n, number of participants in the specific category; TEAEs, treatment-emergent adverse events.
aParticipants are only counted once within each preferred term for the most related occurrence.
fEvent specific to females.
mEvent specific to males.
#Percentages for sex-specific serious TEAEs were calculated relative to the number of female or male participants.
According to the current FDA label for atogepant, the most common adverse reactions (those greater than placebo and with an incidence of at least 2%) were constipation, nausea, fatigue, and decreased appetite (10). We examined the number of adverse events, time to onset, use of concomitant medication to treat the TEAE, duration, severity, and outcome for these four adverse reactions (Table 6). For constipation, there were 24 events reported in 23 participants, with a median time to onset of 17 days (range of 2–163 days; mean [SD] of 45.5 [54.7] days). All cases of constipation were mild or moderate in severity; no severe cases were reported. One participant discontinued atogepant treatment due to constipation. Median constipation duration was 116 days (range 2–312; mean [SD] 123.0 [110.5]), and 21/24 events resolved or were resolving by the end of the trial. Most of these events were managed with over-the-counter laxative products. Across all four adverse drug reactions, no severe cases were reported. The data for decreased appetite, nausea, and fatigue are shown in Table 6.
Adverse drug reactions characteristics: constipation, nausea, fatigue/somnolence, and decreased appetite.

N, number of participants in the safety population; n, number of participants in the specific category; SD, standard deviation; TEAEs, treatment-emergent adverse events.
aPercent based on number of participants in the safety population (N = 685).
bPercent based on number of TEAE events.
cDuration was calculated for TEAEs with resolution as end date – start date + 1.
dFor one participant who reported nausea outcome as resolved, the TEAE ending date was incomplete (because it only reported month), so that TEAE duration could not be calculated.
eAll fatigue/somnolence data represent pooled data except for severity data, which are noted individually.
Hepatic laboratory values
Post-baseline hepatic-related parameters of clinical interest are listed in Table 7. Of the 683 participants who had at least one post-baseline value for alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and total bilirubin (TBIL), four (0.6%) participants had an ALT or AST ≥3 times the upper limit of normal (ULN, Table 7). The aminotransferase values for these four participants resolved without additional treatment, and none of the four participants presented with liver disease-associated symptoms. One of the four participants with an elevated aminotransferase at Visit 1 was withdrawn from the trial, as required by trial protocol when aminotransferase increases are observed at screening. The other three participants with elevated aminotransferase values resolved as they continued atogepant treatment (as their aminotransferase increases were observed after Visit 1). The external Clinical Adjudication Committee formed by an independent panel of liver experts who assessed causality using a 3-category scale (Probable, Possibly or Unlikely) determined that elevations in aminotransferase in two participants were unlikely related to atogepant treatment; elevations in the other two participants were considered possibly related to atogepant treatment (online Supplemental Table S5). No participants in the open-label extension trial met Hy's Law criteria (Table 7).
Post-baseline hepatic-related parameter values of clinical interest (safety population).

ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; N, number of participants in the safety population; N1, number of participants with at least one non-missing post-baseline value; n, number of participants in the specific category; TBIL, total bilirubin; ULN, upper limit of normal value.
aPercentages are calculated as 100 × (n / N1).
Columbia Suicide Severity Rating Scale (C-SSRS)
While the C-SSRS demonstrated that 678 (99.0%) participants had no suicidal ideation, one (0.1%) participant had a “non-specific active suicidal thought”, and three (0.4%) participants expressed a “wish to be dead”. However, no suicidal behaviors were noted, with 682 (99.6%) of the safety population not reporting suicidal behavior per the C-SSRS.
Additional outcomes assessed
Mean changes from baseline for systolic and diastolic blood pressure and pulse rate increased approximately 1 mmHg and 1 beat per minute, respectively. None of the 683 participants had a “potentially clinically significant” increase in sitting systolic blood pressure during the trial (potentially clinically significant, high: an observed value ≥180 mmHg or a change from baseline increase of ≥20 mmHg); seven (1%) participants had a potentially clinically significant elevation in sitting diastolic blood pressure (potentially clinically significant, high: an observed value ≥105 mmHg or a change from baseline increase of ≥15 mmHg;). A potentially clinically significant decrease in weight of ≥7% at any point during the treatment period was seen in 163 of 683 (23.9%) participants, while 63 (9.2%) experienced a potentially clinically significant increase in weight of ≥7% at any point during the treatment period. The mean change in body weight from baseline to the end of treatment was −1.77 kg. Most participants with normal baseline ECG remained within normal limits at the end of the trial (454 out of 542, 83.8%; online Supplemental Table S6). On day 280, one participant had a TEAE of atrioventricular block first degree, which was considered mild and not related to atogepant treatment by the investigator (1 out of 542, 0.2%; online Supplemental Table S6).
Discussion
This multicenter, open-label, 40-week extension (309-OLEX) of the pivotal 12-week ADVANCE phase 3 trial met the primary objective of evaluating the safety and tolerability of long-term oral atogepant 60 mg once daily treatment. The results presented here are consistent with the safety profile of atogepant in previous placebo-controlled, double-blind, randomized trials (11,12). In the 309-OLEX trial, there were no new safety signals identified, and no deaths occurred over the course of the trial. More than 70% of the safety population continued atogepant treatment ≥9 months, consistent with reported tolerability in previous trials (11,12).
A total of 685 participants who completed the 12-week phase 3 ADVANCE trial received at least one dose of oral atogepant 60 mg once daily in the 40-week 309-OLEX trial. In this trial, the mean exposure to atogepant was 233.6 days, with over 50% of the safety population exposed to atogepant for ≥280 days. Discontinuations in atogepant-treated participants due to lack of efficacy (n = 4) or adverse events (n = 25) were low (0.6% and 3.6%, respectively); 2.6% (n = 18) of discontinuations were due to restrictions related to COVID-19.
The overall incidence of TEAEs in this 40-week trial was similar to that seen in the previous 12-week trials (phase 2b/3 and ADVANCE trials). TEAEs for atogepant 60 mg in the 309-OLEX trial (63%) were similar to atogepant 60 mg in the phase 2b/3 and ADVANCE trials, (58% and 54%, respectively) (11,12). Although the current trial did not include a placebo arm, the overall TEAE incidence in the placebo arms of the phase 2b/3 and ADVANCE trials were 49% and 57%, respectively (11,12). TEAEs for atogepant 30 mg in the ADVANCE trial (52%) (12) and for atogepant 10 mg in the phase2b/3 trial (66%) (11) were similar to those seen in the current trial (63%).
The TEAEs occurring in ≥3% of safety population participants in the current trial were upper respiratory tract infection, urinary tract infection, nasopharyngitis, sinusitis, as well as constipation and nausea, similar to what was observed in the phase 2b/3 and ADVANCE trials (11,12). TEAEs considered to be treatment-related ranged from 18% to 26% in the phase 2b/3 trial (11) and from 15% to 23% in the ADVANCE (12) atogepant arms of the 12-week trials. In contrast, this 40-week trial observed a relatively lower rate of treatment-related AEs (9%), with 59% of TEAEs considered mild to moderate in severity. Constipation and nausea were common treatment-related AEs in this trial, consistent with the previously reported 12-week trials (11,12). Potentially clinically significant weight loss was more common than weight gain. Serious TEAEs were low across all three trials (11,12), with only one serious TEAE considered treatment-related by the investigators (12), and none observed in the 309-OLEX trial.
Because first generation gepants (small-molecule CGRP receptor antagonists) were implicated in potential drug-induced liver damage (14,15), ALT and AST levels are closely monitored in gepant trials. A small number of participants with elevations of ALT or AST ≥3 times the upper limit of normal have been observed in both the 12-week (11,12) and the current trial, but no overall hepatic safety issues in any of the trials (11,12) were identified. No Hy’s Law cases have been reported.
Preventive treatment of migraine necessitates medications with favorable long-term benefit-risk profiles, as migraine is a disease that often persists throughout a person's lifetime (8). Therefore, long-term safety and tolerability studies of preventive migraine medications are needed. Long-term trials of at least 52-weeks or longer are published for CGRP-inhibiting monoclonal antibodies (mAbs) including framenezumab (16), galcanezumab (17), eptinezumab (18), erenumab (8) and gepants including atogepant (9) and rimegepant (19). The percentage of participants experiencing at least one TEAE ranged from 71% to 89% (depending on treatment arm investigated) in mAb trials (8,16–18), with the current 309-OLEX (gepant) trial demonstrating a relatively lower rate of 63%, similar to that observed in another gepant trial (19). The lack of injection site reactions may account for the lower rate of TEAEs with gepants. Upper respiratory tract infection, nasopharyngitis, and sinusitis were common TEAEs in the current trial as well as in the mAbs and gepant trials referenced above (16–19). The incidence of upper respiratory tract infection observed in the 309-OLEX trial (5.5%), which was not considered treatment-related, is similar to the incidence of upper respiratory tract infection observed in the placebo arms of the two atogepant 12 week trials (8% and 4.5%, respectively) (11,12). TEAEs leading to discontinuation in the current trial (3.2%) were also consistent with mAbs and gepant trials, where TEAEs leading to discontinuation ranged from 2% to 6%, depending on the treatment arm assessed (8,16–19). All 6 trials, and the current trial, concluded that their respective CGRP-antagonists were safe and well tolerated over the duration of their respective treatment periods, which ranged from 52 weeks up to five years (8,9,16–19). The safety data in this trial was consistent with that of the 52-week atogepant trial previously reported (9).
Despite the 309-OLEX trial having met its primary objective of assessing safety and tolerability of long-term (40-week) atogepant treatment, the current trial has limitations. For example, no standard care control arm was incorporated into the trial design, limiting the extent to which the atogepant-treated participants can be compared to participants using other non-gepant usual care modalities for 40 weeks. The 52-week atogepant trial had a standard care control arm to provide context for the comparison of TEAEs (9). Since this trial was focused on safety and tolerability, efficacy was not measured, so no conclusions around efficacy can be drawn. However, four (0.6%) participants discontinued treatment in the open-label treatment period due to lack of efficacy. One additional limitation is that the study excluded participants with substantial comorbid disorders of the cardiovascular, endocrine, gastrointestinal, hematologic, hepatic, neurologic, pulmonary, or renal systems as well as those with hypertension (as defined by a sitting systolic blood pressure >160 mmHg or a sitting diastolic blood pressure >100 mmHg), ECG abnormalities, or women who were pregnant or planning to become pregnant. Furthermore, this trial prohibited the concomitant use of warfarin, erythromycin, and topiramate as well as other medications (see online Supplemental Table S3 for full list). The safety findings presented here were observed in this clinical trial population.
Given the pathophysiology of CGRP in migraine and cardiovascular disease, concerns regarding cardiovascular safety of CGRP inhibitors for migraine treatment remain, despite the relatively low incidence of cardiovascular adverse events reported in CGRP-inhibiting trials (20,21). As the 40-week 309-OLEX trial is relatively short to adequately assess long-term hemodynamic effects, a more comprehensive post-hoc analysis of cardiovascular risk factors in the atogepant-treated population is needed.
In conclusion, this 40-week, open-label extension of the ADVANCE trial demonstrated that the safety and tolerability of long-term atogepant treatment are consistent with the pivotal 12-week, randomized, placebo-controlled phase 2b/3 and phase 3 trials. No safety signals were identified in the 309-OLEX trial. Overall, the data from this long-term extension trial is consistent with the known safety profile for atogepant and further supports once daily dosing of oral atogepant 60 mg up to 40 weeks.
Article highlights
This long-term open-label extension trial demonstrated that atogepant safety and tolerability aligns with the safety profiles from the pivotal phase 3 trials. No deaths and no previously unidentified safety issues were seen. The data from this extension trial supports once daily dosing of oral atogepant 60 mg as safe and tolerable in trial participants.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024221128250 - Supplemental material for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial
Supplemental material, sj-pdf-1-cep-10.1177_03331024221128250 for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial by Brad C Klein, Rosa Miceli, Lawrence Severt, Peter McAllister, Laszlo Mechtler, Jennifer McVige, Merle Diamond, Michael J Marmura, Hua Guo, Michelle Finnegan and Joel M Trugman in Cephalalgia
Supplemental Material
sj-pdf-2-cep-10.1177_03331024221128250 - Supplemental material for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial
Supplemental material, sj-pdf-2-cep-10.1177_03331024221128250 for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial by Brad C Klein, Rosa Miceli, Lawrence Severt, Peter McAllister, Laszlo Mechtler, Jennifer McVige, Merle Diamond, Michael J Marmura, Hua Guo, Michelle Finnegan and Joel M Trugman in Cephalalgia
Supplemental Material
sj-pdf-3-cep-10.1177_03331024221128250 - Supplemental material for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial
Supplemental material, sj-pdf-3-cep-10.1177_03331024221128250 for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial by Brad C Klein, Rosa Miceli, Lawrence Severt, Peter McAllister, Laszlo Mechtler, Jennifer McVige, Merle Diamond, Michael J Marmura, Hua Guo, Michelle Finnegan and Joel M Trugman in Cephalalgia
Supplemental Material
sj-pdf-4-cep-10.1177_03331024221128250 - Supplemental material for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial
Supplemental material, sj-pdf-4-cep-10.1177_03331024221128250 for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial by Brad C Klein, Rosa Miceli, Lawrence Severt, Peter McAllister, Laszlo Mechtler, Jennifer McVige, Merle Diamond, Michael J Marmura, Hua Guo, Michelle Finnegan and Joel M Trugman in Cephalalgia
Supplemental Material
sj-pdf-5-cep-10.1177_03331024221128250 - Supplemental material for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial
Supplemental material, sj-pdf-5-cep-10.1177_03331024221128250 for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial by Brad C Klein, Rosa Miceli, Lawrence Severt, Peter McAllister, Laszlo Mechtler, Jennifer McVige, Merle Diamond, Michael J Marmura, Hua Guo, Michelle Finnegan and Joel M Trugman in Cephalalgia
Supplemental Material
sj-pdf-6-cep-10.1177_03331024221128250 - Supplemental material for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial
Supplemental material, sj-pdf-6-cep-10.1177_03331024221128250 for Safety and tolerability results of atogepant for the preventive treatment of episodic migraine from a 40-week, open-label multicenter extension of the phase 3 ADVANCE trial by Brad C Klein, Rosa Miceli, Lawrence Severt, Peter McAllister, Laszlo Mechtler, Jennifer McVige, Merle Diamond, Michael J Marmura, Hua Guo, Michelle Finnegan and Joel M Trugman in Cephalalgia
Footnotes
Acknowledgements
AbbVie and the authors thank the participants, trial sites, and investigators who participated in this clinical trial. AbbVie Inc. (formerly Allergan) funded this trial and participated in the trial design, research, analysis, data collection, interpretation of data, review, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship. Medical writing support was provided by Matthew R Distasi, of AbbVie Inc., and editorial support was provided by Angela T Hadsell of AbbVie Inc, both funded by AbbVie Inc. The listed authors have authorized submission of their manuscript via third party (Matthew R Distasi of AbbVie Inc., funded by AbbVie Inc.) and have approved any declarations of conflicting interests and funding statements.
Data availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: ![]() .
.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: BCK reports advisory board participation for AbbVie Inc., Amgen, Biohaven, Eli Lilly and Company, Promius, and Lundbeck; speaker/speaker board participation for AbbVie Inc., Amgen, Biohaven, Eli Lilly and Company, Promius, Teva, Pernix Therapeutics, and Theranica; consultant participation for AbbVie Inc., Amgen, Eagalet, Promius, Teva, and Depomed; receipt of grant support for research or education from AbbVie Inc. and Eli Lilly and Company; and receipt of author royalties from AppsbyDocs, LLC. RM is a former employee of AbbVie Inc., was an employee of AbbVie Inc. at the time of study conduct and of manuscript drafting, and may hold AbbVie Inc. stock. LS is a former employee of AbbVie Inc., was an employee of AbbVie Inc. at the time of study conduct and of manuscript drafting, and may hold AbbVie Inc. stock. PMA reports advisory board participation for AbbVie Inc., Aeon, Amgen, Biohaven, Eli Lilly and Company, Lundbeck, Revance, and Teva; speaker/speaker board participation for AbbVie Inc., Aeon, Amgen, Biohaven, Eli Lilly and Company, Lundbeck, Revance, and Teva; and receipt of grant support for research or education from AbbVie Inc., Amgen, Biogen, Biohaven, EMD Serono, Lundbeck, Novartis, and Teva. LM reports speaker/speaker board participation for AbbVie Inc., Allergan (now AbbVie Inc.), Amgen, Inc., Biohaven Pharmaceuticals, Currax Pharmaceuticals, LLC., electroCore, Inc., Impel Pharmaceuticals, Inc., Lundbeck, Novartis, Promius Pharma, LLC., Teva Pharmaceutical Industries Ltd., and Theranica Bio-Electronics, Ltd.; receipt of grant support for research or education from AbbVie Inc., Aeon BioPharma, Alder BioPharmaceuticals, Inc., Allergan (now AbbVie Inc.), Amgen, Inc., Biohaven Pharmaceuticals, Boston Biomedical, Inc., Charlotte's Web, Inc., Delmar Pharmaceuticals (now Kintara Therapeutics), Lundbeck, Novartis, Orbis Pharma, Inc., Teva Pharmaceutical Industries Ltd., The Harry Dent Family Foundation, Inc., and Theranica Bio-Electronics, Ltd. JMV reports advisory board participation for Allergan (now AbbVie Inc.), AbbVie Inc.; speaker/speaker board participation for Allergan (now AbbVie Inc.), AbbVie Inc., Amgen, Avanir, Biohaven, Eli Lilly and Company, Teva, Neurelis, and Theranica; consultant participation for Allergan (now AbbVie Inc.), AbbVie Inc., and Theranica; receipt of grant support for research or education from Allergan (now AbbVie Inc.)/AbbVie Inc., Amgen, Avanir, Biohaven, Currax, Eli Lilly and Company, Novartis, Lundbeck, Teva, Theranica, and the Harry Dent Family Foundation. MD reports advisory board participation for Allergan (now AbbVie Inc.), AbbVie Inc., Lundbeck, Amgen, Assertio Therapeutics, Axsome Therapeutics, Eli Lilly and Company, Supernus Pharmaceuticals, Teva, and Upsher-Smith Laboratories; speaker/speaker board participation for Allergan (now AbbVie Inc.), AbbVie Inc., Lundbeck, Amgen, Assertio Therapeutics, Eli Lilly and Company, Supernus Pharmaceuticals, and Teva; consultant participation for Allergan (now AbbVie Inc.), AbbVie Inc., Lundbeck, Amgen, Eli Lilly and Company, and Teva; and receipt of author royalties from Allergan (now AbbVie Inc.), AbbVie Inc., Biohaven, Lundbeck, and Teva. MJM reports advisory board participation for Theranica; speaker/speaker board participation for Amgen, Eli Lilly and Company, and Novartis; consultant participation for Alder, Lundbeck, Eli Lilly and Company, Supernus, and Theranica; receipt of grant support for research or education from Allergan (now AbbVie Inc.), AbbVie Inc., Teva, and GammaCore; and receipt of author royalties from Demos Medical, Cambridge University Press, and MedLink Neurology. HG is a full-time employee of AbbVie, Inc., and may hold stock/stock options in the company. MF is a former employee of AbbVie Inc., was an employee of AbbVie Inc. at the time of study conduct and of manuscript drafting, and may hold AbbVie Inc. stock. JMT is a full-time employee of AbbVie Inc., and may hold stock/stock options in the company. RM, LS, and MF were employees of AbbVie Inc. at the time of study conduct and of manuscript drafting.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by AbbVie Inc. (formerly Allergan). Employees of AbbVie Inc. participated in the trial design, research, analysis, data collection, interpretation of data, review, approval of the publication, and the decision to submit for publication. Employees of AbbVie Inc. also provided medical writing and editorial support for this manuscript.
No honoraria or payments were made for authorship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.