
Abstract
Background
Calcitonin gene-related peptide release in trigeminovascular system is a pivotal component of neurogenic inflammation underlying migraine pathophysiology. Transient receptor potential channels and voltage-gated KCNQ/Kv7 potassium channels expressed throughout trigeminovascular system are important targets for modulation of calcitonin gene-related peptide release. We investigated the effects of certain transient receptor potential (TRP) channels the vanilloid 1 and 4 (TRPV1 and TRPV4), the ankyrin 1 (TRPA1), and metastatin type 8 (TRPM8), and voltage-gated potassium channel (Kv7) opener retigabine on calcitonin gene-related peptide release from peripheral (dura mater and trigeminal ganglion) and central (trigeminal nucleus caudalis) trigeminal components of rats.
Methods
The experiments were carried out using well-established in-vitro preparations (hemiskull, trigeminal ganglion and trigeminal nucleus caudalis) from male Wistar rats. Agonists and antagonists of TRPV1, TRPV4, TRPA1 and TRPM8 channels, and also retigabine were tested on the in-vitro release of calcitonin gene-related peptide. Calcitonin gene-related peptide concentrations were measured using enzyme-linked immunosorbent assay.
Results
Agonists of these transient receptor potential channels induced calcitonin gene-related peptide release from hemiskull, trigeminal ganglion and trigeminal nucleus caudalis, respectively. The transient receptor potential channels-induced calcitonin gene-related peptide releases were blocked by their specific antagonists and reduced by retigabine. Retigabine also decreased basal calcitonin gene-related peptide releases in all preparations.
Conclusion
Our findings suggest that favorable antagonists of these transient receptor potential channels, or Kv7 channel opener retigabine may be effective in migraine therapy by inhibiting neurogenic inflammation that requires calcitonin gene-related peptide release.
Keywords
Introduction
Transient receptor potential (TRP) channels are non-selective cation channels that play a crucial role in pain signaling pathways including trigeminal nociception in the migraine (1–3). Growing evidence indicates that some members of TRP channels such as the vanilloid 1 and 4 (TRPV1 and TRPV4), the ankyrin 1 (TRPA1), and metastatin type 8 (TRPM8) are involved in migraine pathophysiology and may represent novel targets for headache therapeutics (1,2). These types of TRP channels are widely expressed in peripheral and central components of trigeminovascular system including trigeminal ganglion neurons, trigeminal meningeal afferents, and spinal trigeminal nucleus (1–4). The majority of these trigeminal afferents consist of C fibers containing CGRP, main contributor of migraine (5). The activation of TRPV1 and TRPA1 by specific agonists induces CGRP release from trigeminovascular structures (1,2,4). However, although it has been reported that activation of TRPV4 and TRPM8 led to CGRP release from non-trigeminal tissues (6,7), their relevance to CGRP release from components of trigeminovascular system remain unclear.
Voltage-gated KCNQ/Kv7 (Kv7.1-Kv7.5) potassium channels, a subfamily of voltage-gated K+ channels, stabilise membrane excitability through non-inactivating K+ currents, and thus contribute to control of neuronal excitability by suppressing membrane depolarisation (8). These channels, particularly Kv7.2 and Kv7.3, are expressed in meningeal afferents, trigeminal ganglion neurons and brainstem that are strategical sites of pain pathway underlying migraine headache (8–10).
Thus, we hypothesise that activation of Kv7 channels by retigabine, an activator for Kv7.2-5, reduces CGRP release from the peripheral and central components of trigeminovascular system stimulated by activation of migraine-related TRP channels (TRPV1, TRPV4, TRPA1 and TRPM8). We therefore explored effects of retigabine on the in-vitro CGRP release induced by agonists of these four TRP channels in peripheral (dura mater and trigeminal ganglion) and central (TNC) trigeminal components of rats.
Materials and methods
Animals
All experimental procedures were approved by Bolu Abant Izzet Baysal University Animal Experiments Local Ethics-Committee (No: 2017/09 and 2020/21). Experiments were performed in male Wistar-rats (210–290 g; 8–10 weeks of age). Rats were purchased from animal breeding-centre of University of Bolu Abant Izzet Baysal, Turkey. Rats were kept in compliance with the National Institutes of Health guide for the care and use of Laboratory animals, and were harboured in their individual cages with a 12-h light/dark cycle at 22 ± 2°C. Rats were allowed to access ad libitum a standard rodent diet and water.
Drugs and reagents
Capsaicin (Cat.#M2028), 4α-Phorbol12,13-didecanoate (4α-PDD,Cat.#P8014), Cinnamaldehyde (Cat.#W228613), Menthol (Cat.#M2772), Capsazepine (Cat.#C191), GSK-2193874 (Cat.#SML0942), HC-030031 (Cat.#H4415), AMTB (Cat.#SML0103), Retigabine (Cat.#SML0325), cOmplete™ protease-inhibitor-cocktail (Cat.#11697498001) and Dimethyl-Sulfoxide (DMSO, Cat.#20-139) were purchased from Sigma-Aldrich (Schnelldorf, Germany). Rat CGRP ELISA kits (Cat.#E-EL-R0135) were purchased from ELABscience (Wuhan, China). All drugs were dissolved in DMSO to obtain stock solutions at 10 mM concentration, then further diluted with synthetic interstitial fluid (SIF) to reach their final concentrations. Maximal DMSO concentration was 3%, and an equivalent volume of SIF containing 3% DMSO was used as vehicle control.
CGRP release experiments from the trigeminovascular system
Sample size in experimental groups was calculated using the G*Power software (version 3.1.9.4). (Supplementary File) A total of 108 rats were used for the experiments. All rats were randomly allocated into the different experimental groups. Moreover the investigator performing the CGRP release experiments was blind to the test compounds administered to the preparations. Two hemiskull and trigeminal ganglion preparations were obtained from each rat while one TNC preparation was obtained.
Nine general groups were established to study each preparation (n = 12 hemiskulls, 6 rats; n = 12 trigeminal ganglia, 6 rats; n = 12 trigeminal nucleus, 12 rats for each group, respectively). Thus, 54 rats were used for hemiskull and trigeminal ganglion preparations, however, since each rat had one brainstem, 54 more rats were added for TNC preparations, unlike the other groups. CGRP release experiments were performed using in-vitro hemiskull, trigeminal ganglion and TNC preparations that were previously well described by us and the other groups (11–14). Shortly, the rats were decapitated following anaesthesia with a mixture of 70% CO2 and 30% O2. Cerebellum was removed, and brainstem was dissected. The TNC running caudally from approximately 13–16 mm from the bregma was then harvested from the brainstem. Then, the skull was cut mid-sagittally and brain halves were rapidly removed while cranial dura mater was left attached to the skull. Then, trigeminal ganglia were collected via dissecting 1 mm proximal and distal to branching point of mandibular nerve, and the dura mater was carefully skinned from the ganglia.
Each of these preparations was immersed and superfused for 30 min with 500 mL of carbogen-gased synthetic interstitial fluid (SIF) of following constituent (mM): NaCl (108), KCl (3.48), MgSO4 (3.5), NaHCO3 (26), NaH2PO4 (11.7), CaCl2 (1.5), sodium-gluconate (9.6), glucose (5.55), and sucrose (7.6), pH 7.4, at 37°C.
Similar to the randomisation procedure for assigning rats to different groups as mentioned above, all preparations obtained were randomised. After washing period, control or test substances were applied to the preparations, as follows: SIF (for basal CGRP measurement), vehicle (control, 3% DMSO), TRPV1 agonist capsaicin (100 nM), TRPV4 agonist 4α-PDD (100 µM), TRPA1 agonist cinnamaldehyde (300 µM), TRPM8 agonist menthol (300 µM), TRPV1 antagonist capsazepine (10 µM), TRPV4 antagonist GSK-2193874 (10 µM), TRPA1 antagonist HC-030031 (50 µM), TRPM8 antagonist AMTB (10 µM), Kv7 channel opener retigabine (10 μM). Each agonist was administered alone or in combination with its antagonist or retigabine in SIF. 200 μl of the sample was collected from the incubation samples at 10-minute intervals and to prevent CGRP degradation samples were immediately transferred to a tube containing 50 μL of protease-inhibitor-cocktail (cOmplete, Sigma-Aldrich). The samples were stored at −20°C until assayed for CGRP immunoreactivities.
Determination of CGRP concentrations in the samples
Basal, control and stimulated CGRP releases were measured in the samples of each preparation. An enzyme-linked immunosorbent assay kit was used to measure CGRP content in the samples, as described previously (15). The detection limit is about 9 pg/ml (ELABscience). Assay procedure was performed in accordance to the instructions of manufacturer and in duplicates. The optical density was measured at 450 nm using a microplate reader (Epoch BioTek Instruments Inc., Winooski, VT, USA). An optical density curve was plotted by using standards with defined CGRP concentrations.
Statistical analysis
Data were given as mean ± standard error of mean. Statistical analysis was performed using SPSS for Windows (IBM_SPSS_Statistics for Windows, Version 22.0, IBM Corp., Armonk, NY, USA). The Shapiro-Wilk test was used to test the normality of data. For the repeated measures of CGRP releases, data that normally distributed were compared using parametric the repeated measures ANOVA (within-subjects ANOVA) followed by Bonferroni post-hoc test while data that were not normally distributed were compared using non-parametric Friedman test (Friedman's two-way ANOVA) followed by Wilcoxon signed-rank test. P values <0.05 was accepted as significant.
Results
Basal CGRP levels in hemiskull, trigeminal ganglion and TNC preparations
There were no significant differences in CGRP levels between baseline (SIF) and control (3% DMSO) treatments in all groups in hemiskull (N = 12), trigeminal ganglion (N = 12) and TNC preparations (N = 12), (P > 0.05 for all groups, Figure 1A–H, Figure 2A–H, Figure 3A–H and Figure 4A–C; Table 1). Additionally, significant difference was not found between basal CGRP levels from the left and right-side parts of all in-vitro preparations in each experimental group (data not shown).

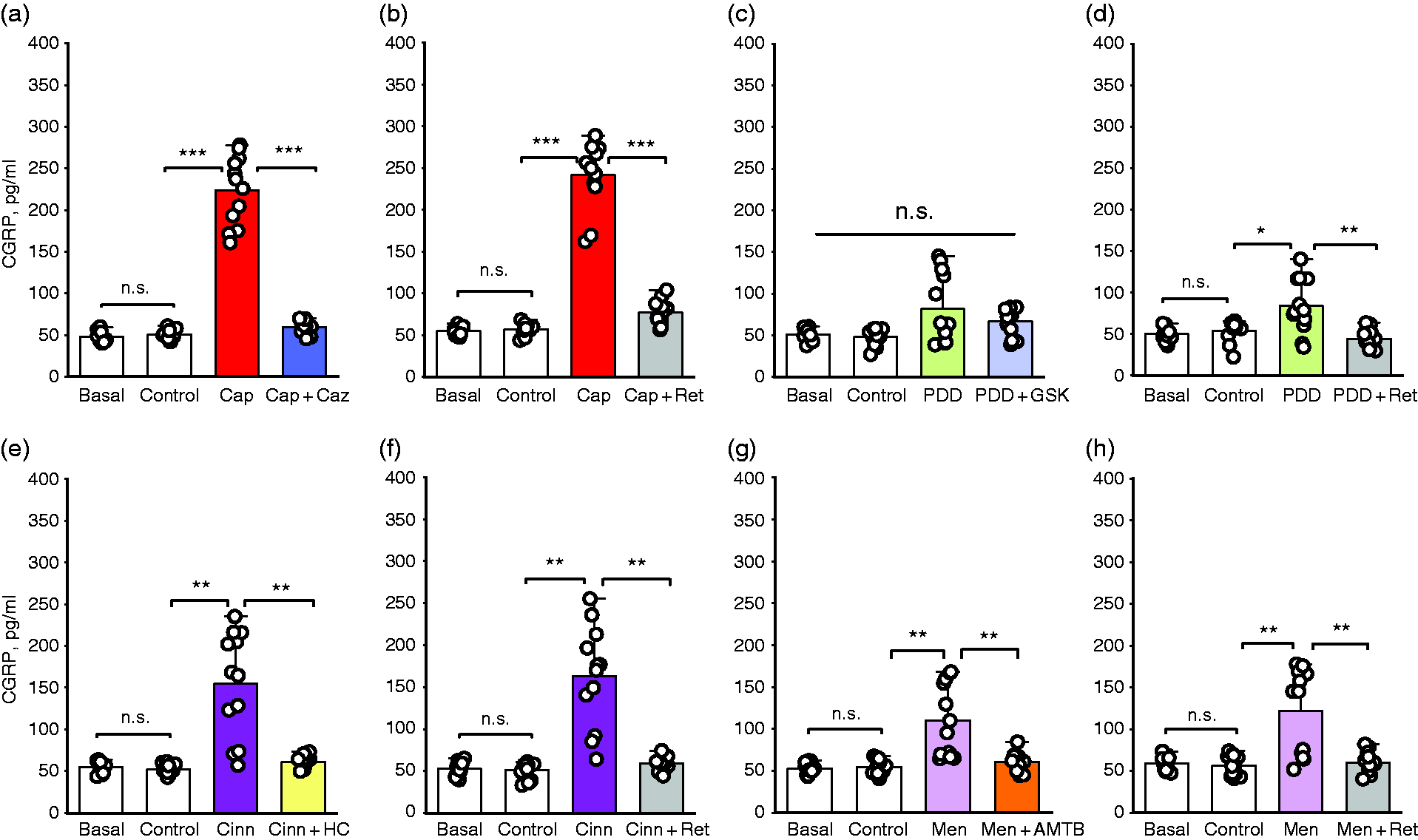
The effects of activation of the TRP and Kv7 channels on the release of CGRP from the hemiskull preparations.
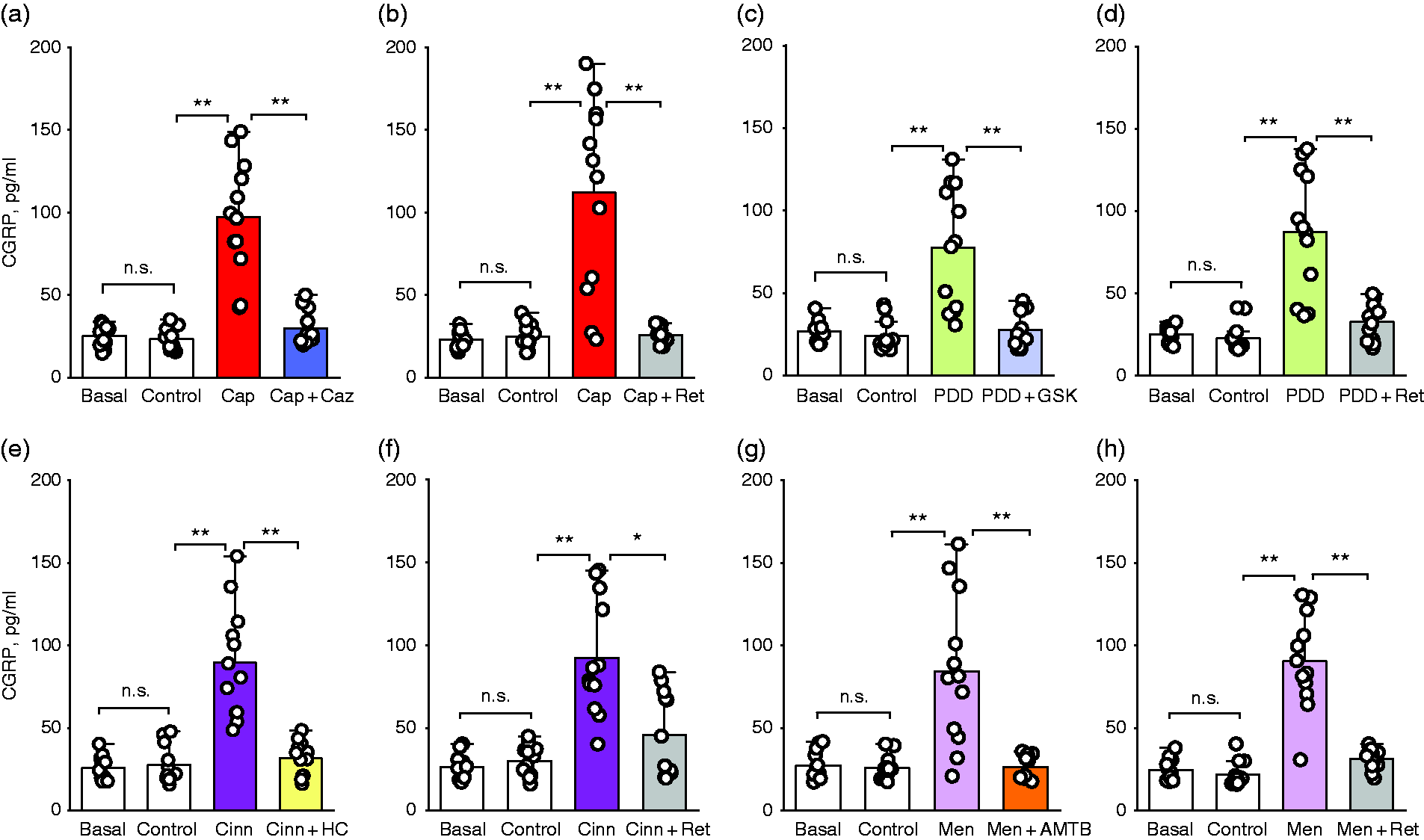
The effects of activation of the TRP and Kv7 channels on the release of CGRP from the trigeminal ganglion preparations.
The effects of activation of the TRP and Kv7 channels on the release of CGRP from the TNC preparations.

The effects of Kv7 channel opener retigabine on the basal CGRP release from the trigeminovascular system and comparison of the CGRP release-inducing effects of the TRP agonists.
Statistical values of calcitonin gene-related peptide releases influenced by the transient receptor potential (TRP) agonist, antagonist and retigabine treatments.

Normally distributed groups were analyzed by RM-ANOVA (repeated measures ANOVA) while non-normally distributed groups were analyzed by Friedman test (Friedman's two-way ANOVA).
Cap, capsaicin; Caz, capsazepine; Cinn, cinnamaldehyde; df, degrees of freedom; HC, HC-030031; Men, menthol; PDD, 4α-PDD; Ret, retigabine; RM-ANOVA, repeated measures ANOVA.
*There is a significant difference between treatments in the respective group.
Effects of TRP channels modulation and Kv7 channel opener retigabine on CGRP release in hemiskull preparations
Capsaicin (P = 0.0001, N = 12), 4α-PDD (P = 0.002, N = 12), cinnamaldehyde (P = 0.0001, N = 12), and menthol (P = 0.006, N = 12) significantly stimulated CGRP release from trigeminal meningeal afferents compared to their controls, respectively (relevant agonist versus its control, respectively, Figure 1A, C, E, G; Table 1 and Supplementary Table 1).
CGRP releases stimulated by the TRP agonists were inhibited by specific TRP antagonists capsazepine (P = 0.0001), GSK-2193874 (P = 0.003), HC-030031 (P = 0.001), and AMTB (P = 0.012), respectively (Figure 1A, C, E and G; Table 1 and Supplementary Table 1).
In a separate set of hemiskull preparations, however, CGRP releases induced by these TRP agonists were significantly diminished by Kv7 channel opener retigabine, respectively (P = 0.0001 for capsaicin vs capsaicin + retigabine; P = 0.0001 for 4α-PDD vs 4α-PDD + retigabine; P = 0.0001 for cinnamaldehyde vs cinnamaldehyde + retigabine; P = 0.0001 for menthol vs menthol + retigabine, N = 12 for each group, respectively, Figure 1B, D, F and H; Table 1 and Supplementary table 1).
Effects of TRP channels modulation and Kv7 channel opener retigabine on CGRP release in trigeminal ganglion preparations
Capsaicin (P = 0.0001, N = 12), cinnamaldehyde (P = 0.001, N = 12) and menthol (P = 0.002, N = 12) significantly induced CGRP release from trigeminal ganglion preparations compared to their controls, respectively (relevant agonist versus its control, respectively, Figure 2A, E and G; Table 1 and Supplementary Table 2).
Although 4α-PDD increased CGRP release from the trigeminal ganglion, this increase was not statistically significant (P = 0.117, N = 12; Figure 2C; Table 1 and Supplementary Table 2).
CGRP releases induced by these three TRP agonists in the trigeminal ganglion preparations were blocked by specific TRP antagonists capsazepine (P = 0.0001), HC-030031 (P = 0.001), and AMTB (P = 0.003), respectively (Figure 2A, E and G; Table 1 and Supplementary table 2).
In a separate set of the trigeminal ganglion preparations, however, CGRP releases induced by four TRP agonists were significantly decreased by Kv7 channel opener retigabine, respectively (P = 0.0001 for capsaicin vs capsaicin + retigabine; P = 0.004 for 4α-PDD vs 4α-PDD + retigabine; P = 0.002 for cinnamaldehyde vs cinnamaldehyde + retigabine; P = 0.004 for menthol vs menthol + retigabine, N = 12 for each group, respectively Figure 2B, D, F and H; Table 1 and Supplementary Table 2).
Effects of TRP channels modulation and Kv7 channel opener retigabine on CGRP release in TNC preparations
Capsaicin, 4α-PDD, cinnamaldehyde, and menthol significantly stimulated CGRP release from the TNC preparations compared to their controls (P = 0.002 and N = 12 for each group), respectively (relevant agonist versus its control, respectively, Figure 3A, C, E and G; Table 1 and Supplementary Table 3).
CGRP releases evoked by the TRP agonists in the TNC preparations were prevented by specific TRP antagonists capsazepine (P = 0.002), GSK-2193874 (P = 0.004), HC-030031 (P = 0.002), and AMTB (P = 0.004), respectively (Figure 3A, C, E and G; Table 1 and Supplementary Table 3).
In a separate set of the TNC preparations, CGRP releases evoked by the TRP agonists were significantly reduced by Kv7 channel opener retigabine, respectively (P = 0.002 for capsaicin vs capsaicin + retigabine; P = 0.005 for 4α-PDD vs 4α-PDD + retigabine; P = 0.023 for cinnamaldehyde vs cinnamaldehyde + retigabine; P = 0.003 for menthol vs menthol + retigabine, N = 12 for each group, respectively, Figure 3B, D, F and H; Table 1 and Supplementary Table 3).
Effects of Kv7 channel opener retigabine alone on basal CGRP releases in hemiskull, trigeminal ganglion and TNC preparations
In order to determine the effects of retigabine on basal CGRP releases in hemiskull, trigeminal ganglion and TNC preparations, we tested retigabine alone in the different sets of these three preparations. Thus, we found that retigabine alone significantly decreased the basal CGRP releases from hemiskull (P = 0.0001, N = 12), trigeminal ganglion (P = 0.002, N = 12), and TNC (P = 0.019, N = 12) preparations compared to their baseline or control values, respectively (Figure 4A–C; Table 2 and Supplementary Table 4A). The effects of activation of the TRP and Kv7 channels on the CGRP release from peripheral and central components of the trigeminovascular system are illustrated in Figure 5.
Statistical values for the effects of retigabine on basal calcitonin gene-related peptide (CGRP) releases and comparison of the effects of the transient receptor potential (TRP) agonists on CGRP releases.

Normally distributed groups were analyzed by RM-ANOVA (repeated measures ANOVA) while non-normally distributed groups were analyzed by Friedman test (Friedman's two-way ANOVA).
df, degrees of freedom; RM-ANOVA, repeated measures ANOVA.
*There is a significant difference between treatments in the respective group.

Schematic representation that depicts the effects of activation of the TRP and Kv7 channels on the release of CGRP from peripheral and central components of the trigeminovascular system.
Different effects of the TRP agonists in inducing CGRP release in hemiskull and trigeminal ganglion, but not TNC, preparations
The amount of TRPV1-mediated CGRP release was significantly greater than that of the other three TRP channels in hemiskull and trigeminal ganglion preparations, respectively (P = 0.0001, Figure 4D and E, Table 2 and Supplementary Table 4B). However, there was no significant difference among the amounts of CGRP induced by the agonists in TNC preparations (N = 24, P = 0.325, Figure 4F; Table 2 and Supplementary Table 4B).
Discussion
Increasing evidence suggests that TRPV1, TRPV4, TRPA1 and TRPM8 of TRP channels are closely related to migraine pathobiology (1,2,16). Previous studies demonstrated that activation of TRPV1 and TRPA1 triggered CGRP release from trigeminovascular structures, respectively (1,2,4). Consistent with those previous studies, in the current study, activation of TRPV1 and TRPA1 by their specific agonists capsaicin and cinnamaldehyde induced CGRP release from the peripheral and central components of trigeminovascular system, respectively. Inhibition of CGRP release by specific antagonists of TRPV1 and TRPA1 further confirmed that these channels stimulate CGRP release in these migraine-related structures.
Additionally, TRPV1 activation was previously shown to induce CGRP release from trigeminal nerve terminals and CGRP-dependent vasodilatation and subsequent neurogenic inflammation within the meninges which are considered an origin site of migraine pain (17,18). Thus, the previous studies clearly indicate that TRPV1-mediated CGRP release may have a role in initiation and propagation of migraine pain. This is also supported by increased TRPV1 expression in chronic migraineurs (19) and increased TRPV1 expression in trigeminal ganglia in preclinical studies (20). However, TRPV1 antagonism alone may not be sufficient for migraine treatment due to clinical trial failure of TRPV1 antagonist SB-705498 (21). However, it should also be taken into consideration that properties of SB-705498 may not be representative of all TRPV1 antagonists.
In the current study, activation of TRPV4 and TRPM8 by specific agonists, 4α-PDD and menthol, also stimulated release of CGRP from these strategical structures of the trigeminovascular system, and the stimulated releases were inhibited by specific antagonists of TRPV4 and TRPM8, respectively. It was previously demonstrated that activation of TRPV4 and TRPM8 caused CGRP release from non-trigeminal tissues (6,7). However, the effects on CGRP release from components of trigeminovascular system were not studied. We revealed that activation of TRPV4 and TRPM8 also induced CGRP release from trigeminal meningeal afferents, trigeminal ganglion and TNC. Our findings are consistent with the fact that these channels are cation channels (fairly high permeability to Ca2+) and lead to neurotransmitter exocytosis through Ca2 + influx. Moreover, the majority of trigeminal afferents consist of C fibers containing CGRP (5).
It has been suggested that TRPV4 and TRPM8 channels are involved in migraine headache, however exactly how they might contribute to migraine remains unclear. A few experimental studies on relevance of TRPM8 in migraine so far have investigated its expression in trigeminovascular system (22,23), but its function and mechanisms still remain unclear. It was shown that activation of meningeal TPRV4 by hypotonic solution or 4α-PDD led to migraine-related pain behavior that was blocked by the TRPV4 antagonist RN1734 in rats (24). It is suggested that increased mechano-sensitivity/stimulation of TRPV4 on the dural afferents may contribute to the throbbing (pulsating) character of migraine pain, because of mechano-stimulation during rapid head movements and coughing are able to worsen headache in migraine sufferers (1). However, role of neither TRPV4 nor TRPM8 in CGRP release from trigeminovascular system has been previously investigated. Thus, our findings suggest that in addition to TRPV1 and TRPA1, TRPV4 and TRPM8 contribute to migraine pathophysiology by triggering CGRP release from the trigeminovascular system structures.
In the current study, activation of TRPV1 induced more CGRP release than activations of the other three TRP channels in the hemiskull and trigeminal ganglion, but not TNC, preparations. There may be several possible explanations for greater amount of TRPV1-mediated CGRP release: i) CGRP-immunoreactive neurons in hemiskull and trigeminal ganglion may colocalise with more TRPV1 immunoreactivity than the other three TRP channels (1,2,10). It has recently been reported that 70% of CGRP-immunoreactive neurons were found to be colocalised with TRPV1 channels in rat dura mater (2). This is also consistent with the meninges being considered as the origin site of migraine pain. ii) The TRPV1 channel may be more sensitive to its agonist capsaicin compared to sensitivities of the other three TRP channels to their agonists. Possible differences in specificities of agonists and antagonists chosen to target TRP channels are also worth discussing. The agonists and antagonists we used are generally considered to be selective and potent for these channels. However icilin, another TRPM8 activator, is more potent than menthol. But we did not choose icilin as it activates not only TRPM8 but also TRPA1. In future, investigations comparing effects of available agonists and antagonists for each of these TRP channels would be useful.
CGRP is released from trigeminal afferents through mechanism of Ca2 + -dependent exocytosis that requires activation of voltage-gated Ca2 + channels in axon terminal by action potential. Voltage-gated Kv7 channels are key physiological regulators of action potentials in nociceptive afferent neurons. Since opening of K+ channels causes hyperpolarisation of membrane potential, such a stabilisation of membrane potential via K+ outflow can limit Ca2 + -dependent exocytosis of CGRP from peptidergic trigeminal afferent nerves. Thus, Kv7 channels may be favorable targets to inhibit nociceptive transmission in migraine-related trigeminal pain pathway.
In the current study, retigabine significantly decreased CGRP releases induced by TRPV1, TRPV4, TRPA1 and TRPM8 from the trigeminovascular system structures, respectively. It also significantly reduced basal CGRP release from those migraine-related structures, respectively. It has been well documented that Kv7 channel openers attenuated hyperalgesias, trigeminal neuropathic and inflammatory pain, and some neuronal hyperexcitation-related conditions in different experimental models (8,25,26). Of the Kv7 channel openers, retigabine was most widely studied to investigate possible therapeutic effects of Kv7 channels activation on these conditions. However, only one study so far explored effects of retigabine on CGRP release from just the brainstem. In that study, it was shown that retigabine inhibited capsaicin-induced CGRP release from rat brainstem explants (27). Our findings are in line with that study, however we provide more and novel evidence for therapeutic effects of retigabine on the basal and stimulated CGRP release from the migraine-related structures. Additionally, our findings are important because of retigabine reduced basal and stimulated CGRP release from not only TNC but also trigeminal ganglion and trigeminal meningeal afferents.
Furthermore, our findings have clinical relevance because retigabine is a drug approved for clinical use in patients with epilepsy as it exerts its effectiveness by suppressing seizure activity (28). It has recently been demonstrated that retigabine dose-dependently inhibited cortical spreading depression (CSD), an underlying phenomenon of migraine aura, in mouse cortical slices (29). Our findings are also consistent with this recent study. Considering that the common mechanism in CSD and epileptic seizure is abnormal neuronal depolarisation and the clinical effectiveness of retigabine for epilepsy patients, the potential for retigabine to be used in migraine treatment in the future may be plausible.
Additionally, in the current study, retigabine reduced basal CGRP releases from the trigeminovascular system structures. The meninges are densely innervated by trigeminal afferent fibers, and considered as the origin site of migraine pain. Pain impulses originating in the meninges are transmitted to second-order neurons in TNC. Thus, inhibition of CGRP release from these two strategic structures is important for migraine treatment. There is a basal CGRP release as a consequence of intrinsic activity of trigeminal sensory neurons. This may be essential for normal physiological functioning. The amount reduced by retigabine in basal CGRP release relative to that in stimulated CGRP release may not be significant for migraine treatment. However, it is known that plasma CGRP levels increase not only during migraine attacks but also between attacks (30). Thus, retigabine may be a promising agent for both prophylactic and symptomatic treatment of migraine due to its reducing effect on CGRP release from the trigeminovascular system.
New migraine-specific drugs targeting the CGRP pathway have been shown to be effective in the acute treatment of migraine (31). These are small-molecule CGRP receptor antagonists (gepants), and monoclonal antibodies targeting either CGRP or its receptor. There are the four FDA approved monoclonal antibodies (erenumab, fremanezumab, galcanezumab and eptinezumab) and the two FDA approved small-molecule antagonists (rimegepant and ubrogepant), which have undergone extensive safety studies (31). It has been reported that gepants provide at least a 50% reduction in monthly migraine days in approximately half of migraine patients (32). However, inhibition of CGRP release from trigeminovascular system by specific TRP antagonists and/or retigabine could pose a potential future target for further reduction of monthly migraine days, or for migraine sufferers for whom these drugs do not work.
Retigabine is a non-selective activator for Kv7.2-5 (8). Here, it may be asked which subtype of Kv7 channels mediates the reduction of CGRP release in trigeminovascular system. Kv7.2 and Kv7.3 of Kv7 channels are predominantly expressed in trigeminovascular system (8–10). Additionally, in the study mentioned above (29) it was demonstrated that Kv7.2 channel activation inhibited the CSD. We therefore speculate that retigabine may exert its CGRP-reducing effects substantially through activation of Kv7.2 and/or Kv7.3. However, in future studies, it would be helpful to investigate effects of selective antagonists to elucidate which subtype of Kv7 channels mediates such effects. Moreover, in case of a possible drug development targeting Kv7 channels, more a selective channel opener would potentially be more effective and have less side-effects.
Additionally, it is worth mentioning that prevalence of migraine is approximately 3 times higher in women than in men (33). We have recently demonstrated that female gender hormones estrogen and progesterone exhibit different effects on CGRP release from trigeminovascular system (34). In the current study, we used only male rats. However, in future studies, it would be interesting to investigate the effects of the TRP channels and retigabine on CGRP release from trigeminovascular system in female rodents with different hormonal status.
In conclusion, favorable antagonists of TRPV1, TRPV4, TRPA1 and TRPM8 channels may therefore represent possible new candidates for migraine therapy. Moreover, retigabine may also be a promising for treatment of migraine.
Limitations
In the current study, for CGRP stimulation, we administered the TRP agonists before their antagonists and then co-administered the agonist with its antagonist or retigabine. Therefore, it may be queried whether a second stimulation is reproducible for the second CGRP release via the same TRP channel, and as a result, a doubt may arise as to whether the blockade with antagonists is due to CGRP depletion. In previous similar studies, no depletion-related reduction in CGRP release was demonstrated at least up to fourth stimulation with capsaicin or KCl (11,12,14,35). However, if we had administered the TRP antagonists before the combination of agonist plus antagonist, it would have further strengthened our findings.
Article highlights
Activation of TRPV1, TRPV4, TRPA1 and TRPM8 channels by their specific activators stimulated CGRP release from hemiskull, trigeminal ganglion and TNC preparations, respectively. The stimulated CGRP releases were blocked by specific antagonists of those TRP channels, respectively. Kv7 channel opener retigabine significantly diminished both basal and the TRP channels-induced CGRP releases.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024221114773 - Supplemental material for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system
Supplemental material, sj-pdf-1-cep-10.1177_03331024221114773 for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system by Arzu Citak, Erkan Kilinc, Ibrahim Ethem Torun, Seyit Ankarali, Yasar Dagistan and Hamit Yoldas in Cephalalgia
Supplemental Material
sj-pdf-2-cep-10.1177_03331024221114773 - Supplemental material for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system
Supplemental material, sj-pdf-2-cep-10.1177_03331024221114773 for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system by Arzu Citak, Erkan Kilinc, Ibrahim Ethem Torun, Seyit Ankarali, Yasar Dagistan and Hamit Yoldas in Cephalalgia
Supplemental Material
sj-pdf-3-cep-10.1177_03331024221114773 - Supplemental material for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system
Supplemental material, sj-pdf-3-cep-10.1177_03331024221114773 for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system by Arzu Citak, Erkan Kilinc, Ibrahim Ethem Torun, Seyit Ankarali, Yasar Dagistan and Hamit Yoldas in Cephalalgia
Supplemental Material
sj-pdf-4-cep-10.1177_03331024221114773 - Supplemental material for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system
Supplemental material, sj-pdf-4-cep-10.1177_03331024221114773 for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system by Arzu Citak, Erkan Kilinc, Ibrahim Ethem Torun, Seyit Ankarali, Yasar Dagistan and Hamit Yoldas in Cephalalgia
Supplemental Material
sj-pdf-5-cep-10.1177_03331024221114773 - Supplemental material for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system
Supplemental material, sj-pdf-5-cep-10.1177_03331024221114773 for The effects of certain TRP channels and voltage-gated KCNQ/Kv7 channel opener retigabine on calcitonin gene-related peptide release in the trigeminovascular system by Arzu Citak, Erkan Kilinc, Ibrahim Ethem Torun, Seyit Ankarali, Yasar Dagistan and Hamit Yoldas in Cephalalgia
Footnotes
Acknowledgements
We thank Handan Ankarali for her help in performing the statistical analysis.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Bolu Abant Izzet Baysal University Scientific Research Fund (Grant number: 2017.08.02.1212).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.