
Abstract
Abstract
Background
CGAR, a Phase 3b open-label study, evaluated the long-term safety of galcanezumab in patients with cluster headache who completed one of two Phase 3 double-blind studies in chronic or episodic cluster headache.
Methods
Patients (N = 164) received galcanezumab 300 mg subcutaneously up to once a month. Primary endpoint was safety, as assessed by treatment-emergent adverse events, serious adverse events, and suicidality. Other endpoints included discontinuation rates, immunogenicity, efficacy as assessed by the Patient Global Impression of Improvement, and health values.
Results
At baseline, mean (standard deviation) age was 48.3 (9.8) years, 75.0% were men, and 85.4% were white. Treatment-emergent adverse events (n = 119 [72.6%]) were mostly mild-to-moderate, with nasopharyngitis the most commonly reported (22.0%). One of 18 serious adverse events was judged as treatment related (constipation). Two patients (1.2%) reported suicidal ideation. Five patients (3.1%) discontinued due to an adverse event. Eight patients were treatment-emergent anti-drug antibody positive, two of whom were not treatment-emergent anti-drug antibody positive in the parent studies. On the Patient Global Impression of Improvement, ≥81% reported their cluster headache status as very much, much, or a little better at Months 1, 6, and 12. Health value scores generally improved from baseline.
Conclusions
In this open-label study, galcanezumab was generally well tolerated and improved patient-reported cluster headache status.
Introduction
Cluster headache (CH) is one of the Trigeminal Autonomic Cephalalgias in the International Classification of Headache Disorders (3rd edition) (ICHD-3) (1). On the basis of pooled data from epidemiological population-based surveys, the lifetime global prevalence of CH is just over 1 in 1000 (2). People with CH experience attacks of excruciating unilateral pain (1). Untreated CH attacks last 15 to 180 minutes and occur from once every other day to 8 times a day during active cluster periods. The pain is associated with cranial autonomic symptoms and/or restlessness or agitation. In episodic CH, attacks occur during active cluster periods generally lasting from 7 days to 1 year (when untreated) separated by pain-free remission periods lasting at least 3 months, whereas in chronic CH the active cluster period lasts for 1 year or longer without remission or with remission periods lasting <3 months. It should be noted that at the time of the study reported herein, the ICHD-3 diagnostic criteria specified pain-free remission periods lasting at least 1 month for episodic CH and a remission period of <1 month for chronic CH (3).
Two double-blind, placebo-controlled Phase 3 studies of galcanezumab for the treatment of CH have been completed: Study CGAL, an 8-week double-blind treatment phase in patients with episodic CH (N = 106; ClinicalTrials.gov identifier: NCT02397473), and Study CGAM, a 12-week double-blind treatment phase and an optional 12-month open-label treatment phase in patients with chronic CH (N = 237; NCT02438826) (4 –6). In both studies, patients were randomized in a 1:1 ratio to double-blind monthly subcutaneous injections of galcanezumab 300 mg or placebo. In CGAL, among patients with episodic CH, the weekly frequency of CH attacks across weeks 1 to 3 was significantly reduced with galcanezumab compared to placebo (difference of 3.5 attacks; p = 0.04) (5). These results supported the approval of galcanezumab 300 mg by the United States Food and Drug Administration for the treatment of episodic CH (7). However, in the chronic CH study CGAM, the primary efficacy endpoint was not met (the mean change in weekly frequency of attacks was −4.6 for placebo versus −5.4 for galcanezumab; p = 0.334) (4). Adverse events (AEs) during double-blind treatment were generally mild to moderate in severity, with the most commonly reported AEs for galcanezumab (in ≥5% of patients) being injection-site pain and nasopharyngitis in CGAL and injection site pain, nasopharyngitis, injection-site erythema, and nausea in CGAM (4,5).
Here we provide the results of Study CGAR, a prospective, open-label, Phase 3b study that was designed to provide longer-term data on galcanezumab for the treatment of CH in patients who had completed one of the two Phase 3 CH studies described above. Study CGAR did not have a single specified end-of-study definition and implemented a dosing strategy to more closely approximate what may occur in clinical practice to individualize galcanezumab treatment based on the clinical response for each patient. The primary objective of the study was to evaluate the safety of open-label galcanezumab 300 mg through assessment of AEs, including treatment-emergent AEs (TEAEs), serious AEs (SAEs), and suicidal ideation and behaviors. Additional objectives were to evaluate the efficacy of galcanezumab using the Patient Global Impression of Improvement (PGI-I) and other health value measures, to characterize discontinuations due to AEs, and to characterize the immunogenicity of galcanezumab.
Methods
Study design and participants
Participants were ≥18 years of age who had completed all phases of one of the Phase 3, double-blind, placebo-controlled galcanezumab parent studies, CGAL (for patients with episodic CH (5)) or CGAM (for patients with chronic CH (4)). The study designs and inclusion/exclusion criteria of the parent studies have been previously described in detail elsewhere (4,5). Briefly, to be eligible for CGAL/CGAM, patients met ICHD-3 (beta version) (3) diagnostic criteria for episodic or chronic CH and had at least one attack every other day, at least four total attacks, and no more than eight attacks per day during the baseline assessment period. In CGAL, patients also had a history of CH periods lasting at least 6 weeks (5). Complete inclusion and exclusion criteria for CGAR are provided in the Supplemental Appendix. Although preventive medications were not allowed in CGAL and up to six preventives were allowed in CGAM, once enrolled in CGAR, the following preventive medications were allowed: botulinum toxin type A or B, candesartan, clonidine, cyclandelate, gabapentin, lithium, melatonin, topiramate, valproate, and verapamil (maximum daily dose 480 mg) (4,5).
Study CGAR consisted of a screening phase, during which eligibility was reviewed, and an open-label treatment phase, during which all participants received galcanezumab 300 mg administered as three injections of 100 mg/1.0 mL up to once a month as determined by the study investigator on the basis of clinical symptoms and response. The investigator could choose not to dose patients with galcanezumab for one or more months at their clinical discretion. The open-label treatment phase was of an indefinite duration (i.e., until sponsor decision to end the study based on approval or non-approval from a regulatory agency in the country or region where the patient was enrolled).
All participants provided informed consent before study participation, and ethics review boards at each site approved the study protocol and the informed consent form. The study was conducted in compliance with Good Clinical Practice and the Declaration of Helsinki guidelines and is registered at ClinicalTrials.gov (identifier: NCT02797951).
Safety assessments
Safety was assessed using AEs, including TEAEs, SAEs, AEs leading to discontinuation, vital signs and weight, laboratory measures, and electrocardiograms. Adverse events were coded using MedDRA® Version 23.1. Hypersensitivity-related TEAEs that were identified using standard MedDRA® queries were reviewed by a physician to determine whether they were likely hypersensitive in nature. An SAE was defined as any AE that resulted in death; was life-threatening or placed the participant at immediate risk of death from the event as it occurred; required or prolonged hospitalization; caused persistent or significant disability or incapacity; resulted in congenital anomalies or birth defects; or was another condition that the investigator judged to be significant for any other reason. A sustained increased in blood pressure (BP) was defined as meeting high criteria (systolic BP: ≥140 mm Hg and increase from baseline ≥20 mm Hg; diastolic BP: ≥90 mm Hg and increase from baseline ≥10 mm Hg) at two or more consecutive visits. Suicidal ideation and behaviors were also assessed using the Columbia-Suicide Severity Rating Scale (C-SSRS) (8).
Immunogenicity assessment
Blood samples were collected for immunogenicity testing. Galcanezumab treatment-emergent anti-drug antibody (TE ADA) status was determined using a validated affinity capture elution ELISA, coupled with an additional up-front acid pretreatment step as described in the parent studies (4,5). For patients evaluable for TE ADA, TE ADA+ was defined as having either no ADAs at baseline and ≥1 ADA postbaseline with a titer ≥1:20, or having both baseline and postbaseline ADAs, with the postbaseline titer being at least four-fold greater than the baseline titer.
Efficacy assessments
Efficacy was measured using the PGI-I scale, a patient-reported assessment of their CH condition since they started taking the medication (7-point scale, with 1 = “very much better,” 2 = “much better,” 3 = “a little better,” 4 = “no change,” 5 = “a little worse,” 6 = “much worse,” and 7=“very much worse”) (9). Current health status was assessed using the European Quality 5-Dimensions 5-Levels (EQ-5D-5L) Scale, which is a patient-rated scale that includes the following components: 1) responses to problems (i.e., no problems, moderate problems, severe problems, or extreme problems) in 5 dimensions of health (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression); 2) a Health State Index score, which computed quality-adjusted life years for utilization in health economic analyses, including a United Kingdom (UK) population-based index value and a United States (US) population-based index value (scored as a single value on a scale from less than 0 to 1, with negative values representing a health state worse than dead, 0 equivalent to death, and 1 equivalent to perfect health); and 3) the European Quality Visual Analogue Scale (EQ-VAS), whereby patients rated their perceived health state from 0 (the worst health the patient can imagine) to 100 (the best health the patient can imagine) (10,11).
Statistical methods
The sample size for this study was not based on statistical or power considerations; rather, the sample size was determined by the number of patients completing Studies CGAL and CGAM who chose to enroll in this study.
Analyses were conducted in the galcanezumab-treated population, which included all patients who were enrolled and received at least one dose of study drug. Galcanezumab treatment time is composed of monthly dosing intervals, that is, the time from a galcanezumab dose to the next monthly visit. Off-treatment time is the time between the first monthly visit that a dose was not administered and the dosing re-initiation visit.
Safety endpoints were summarized descriptively. Descriptive summaries of the PGI-I were calculated at 1, 6, and 12 months and summarized as both a categorical and continuous variable. Descriptive statistics for the EQ-5D-5L Health State Index overall score based on US and UK population, scores for each of the 5 EQ-5D-5L dimensions (mobility, self-care, usual activities pain/discomfort, and anxiety/depression), and EQ-VAS Current Health Score were calculated by visit. The number and percentage of participants with dichotomized responses (problems, no problems) for each of the 5 EQ-5D-5L dimensions were also calculated by visit. To assess the correlation of EQ-5D-5L Health State Index Scores and EQ-VAS Current Health Score with PGI-I and CH status, the following analyses were performed: a Spearman correlation analysis between mean change from baseline in EQ-5D-5L with PGI-I at Month 1 and an analysis of variance comparing mean change from baseline in EQ-5D-5L by CH status (active or remission) at Months 1 and 6. Last-observation-carried-forward was used in handling of missing data. Statistical analyses were performed using SAS® software.
Results
Patient disposition
CGAR was conducted at 39 study sites in North America (US and Canada) and Europe (Belgium, Denmark, Finland, France, Germany, Greece, Italy, the Netherlands, Spain, and the UK). The first patient was enrolled (assigned to therapy) on 15 August 2016. The study was terminated by the sponsor due to approval of galcanezumab for the treatment of episodic CH in the US and Canada, with last patient visit on 1 October 2019 and 21 January 2021, respectively. For Europe, the study was terminated by the sponsor due to non-approval by the European Medicines Agency, with last patient visit on 30 June 2020.
Of the 183 patients who were screened, 164 received ≥1 dose of galcanezumab, the majority of whom (70.7%) continued in the study until approval or non-approval from their respective regulatory agency (Figure 1).

Flow of study participants. aIncluded one event each of the following adverse events: lung adenocarcinoma, CH, hematospermia, and angioedema. bDeath due to suicide 46 days after receiving the last dose of galcanezumab; considered not treatment or study related by the investigator. cThe study was terminated by the sponsor due to approval of galcanezumab for the treatment of episodic CH in the US (last patient visit: 01 October 2019) and Canada (last patient visit: 21 January 2021), and due to non-approval in Europe (last patient visit: 30 June 2020). Abbreviations: CH = cluster headache.
Baseline characteristics
A majority of patients (79.3%) were ≥40 years of age (mean age [standard deviation] = 48.3 (9.8) years; range, 23–66 years); most (75.0%) were men and most (85.4%) were white (Table 1). The parent study was CGAM (chronic CH) for 67.7% of the patients and CGAL (episodic CH) for 32.3%. Baseline age, gender, race, and ethnicity of patients who enrolled in CGAR from the parent study CGAL were similar to those of patients from CGAM. Overall, 92.1% of patients were taking ≥1 concomitant medication, the most common of which were sumatriptan (45.1%) and oxygen (29.3%) for abortive treatment and verapamil (34.2%) for prophylaxis.
Baseline patient characteristics.

Abbreviations: CH = cluster headache; n = number of patients; SD = standard deviation.
Data reported as n (%) unless otherwise noted.
Safety
The mean duration of exposure was 475 days (range, 28-1211 days), or approximately 16 months. During the study, 148 patients (90.2%) received at least three doses, 118 (72.0%) at least six doses, 88 (53.7%) at least 12 doses, and 42 (25.6%) at least 24 doses. The highest number of full doses received was 39 doses received by one patient. Ninety-six patients went through uninterrupted monthly dosing, defined as consecutive doses from the first dose until patient discontinuation.
A summary of AEs during galcanezumab-treatment and off-treatment time is shown in Table 2. Of the 119 patients (72.6%) who reported ≥1 TEAE, 79.8% reported the event as mild or moderate. Cluster headache was the only TEAE reported by more than one patient as severe (n = 7). The most frequently reported TEAE was nasopharyngitis (22.0%). The most frequently reported TEAEs by system organ class were infections and infestations (n = 78; 47.6%). Of those, one SAE of gastroenteritis was severe (considered not related to study drug; resolved 14 days after it started). Hypertension was reported as a TEAE for eight patients, three of whom had pre-existing hypertension and the remainder had other pre-existing medical conditions (i.e., hyperlipidemia, hypercholesterolemia, and hepatic steatosis). All of the hypertension TEAEs were of mild or moderate severity, and none was considered by the investigator to be related to galcanezumab treatment.
Summary of adverse events during galcanezumab-treatment and off-treatment time.

Abbreviations: AE = adverse event; CH = cluster headache; n = number of patients; SAE = serious AE; TEAE = treatment-emergent AE.
All data reported as n (%).
aAdverse events coded using MedDRA Version 23.1.
bAlso included in “SAEs” and “discontinuations due to an AE.”
cDeath due to suicide 46 days after receiving the last dose of galcanezumab; considered not treatment or study related by the investigator.
Hypersensitivity-related AEs, injection site reactions, and upper respiratory tract infection-related AEs are shown in Table 3. None of these AEs were judged serious, and all were of mild or moderate severity, except for one patient reporting severe angioedema and pruritus allergic (see below for additional information), one patient reporting severe injection site pain (resolved in three days), and one patient reporting severe injection site pruritus (resolved in three days). Only injection site induration, injection site erythema, nasopharyngitis, and sinusitis occurred in more than five patients each, and no participants discontinued due to upper respiratory tract infection-related or injection site-related TEAEs.
Likely hypersensitivity-related adverse events, injection site reactions, and upper respiratory tract infection-related adverse events (occurring in ≥1% of patients) during galcanezumab-treatment and off-treatment time.

Abbreviations: n = number of patients.
All data reported as n (%).
aAdverse events coded using MedDRA Version 23.1.
Seventeen (10.4%) patients reported 18 treatment-emergent SAEs; only CH occurred in more than one patient (n = 3) (CH could be an SAE if it worsened and met SAE criteria) (Table 4). Constipation was considered by the investigator to be related to galcanezumab treatment (pre-existing constipation worsened 20 months after starting treatment; resolved 37 days later on continued treatment).
Serious adverse events during galcanezumab-treatment and off-treatment time.

Abbreviations: n = number of patients; SAE = serious adverse event.
All data reported as n (%).
aAdverse events coded using MedDRA Version 23.1.
bOne patient reported 2 SAEs (gastroenteritis and cluster headache).
cConsidered treatment or study related by the investigator.
One death occurred during the study. This patient died due to suicide 46 days after receiving the last dose of galcanezumab. C-SSRS assessments were negative at all visits. The investigator judged the suicide to be due to severe family trouble and not related to study procedure or galcanezumab treatment. Five patients (3.1%) discontinued due to an AE (one each of lung adenocarcinoma, CH, hematospermia, angioedema, and completed suicide) (Figure 1; Table 2), of which only angioedema was considered by the investigator to be related to galcanezumab treatment (three days after the 23rd monthly dose, the patient reported angioedema and pruritus allergic, which resolved on the same day).
There were no clinically meaningful findings based on changes in laboratory parameters, weight, and temperature and no clinically meaningful changes in heart rate, PR, QRS intervals, or QTc intervals (data not shown). Hypertension was one of the most common pre-existing conditions at baseline (n = 13; 7.9%). Of 153 patients with baseline and postbaseline measurements, 13 (8.5%) experienced a sustained elevation in sitting systolic or diastolic BP during galcanezumab-treated time, with one patient meeting the criteria for both sustained systolic and diastolic BP (Supplemental Table S1). One patient with sustained systolic BP and four of the patients with sustained diastolic BP had pre-existing hypertension conditions. The one patient with both sustained systolic and diastolic BP did not report pre-existing hypertension but had elevated BP values during screening.
Columbia-Suicide Severity Rating Scale (C-SSRS)
Two patients (1.2%) experienced suicidal ideation (“wish to be dead”; one reported during treatment) but did not have emergence of suicidal behavior.
Immunogenicity - Treatment-emergent Anti-drug Antibodies (TE ADA+)
Of 159 patients who were evaluable for TE ADA, eight patients were TE ADA+ postbaseline in CGAR, two of whom had not previously been found TE ADA+ in either the parent study or at CGAR baseline. No hypersensitivity events were reported by patients who met the TE ADA+ criteria in CGAR.
Efficacy
Using the PGI-I, a majority (≥81%) of patients reported the improvement in their CH condition as “very much better (PGI-I = 1),” “much better (PGI-I = 2),” or “a little better (PGI-I = 3)” at Months 1, 6, and 12, and over half (≥56%) of patients reported the improvement as “very much better (PGI-I = 1)” or “much better (PGI-I = 2)” (Figure 2a). The mean PGI-I score was 2.4 at Month 1, 2.0 at Month 6, and 1.8 at Month 12. Similar findings were observed between patients from the parent study CGAL and those from CGAM (Figures 2b and 2c) at Month 1 (improved PGI-I scores: CGAL 80.4%, CGAM 81.9%; mean PGI-I scores: CGAL 2.5, CGAM 2.4), Month 6 (improved PGI-I scores: CGAL 87.0%, CGAM 92.4%; mean PGI-I score: CGAL 2.0, CGAM 1.9), and Month 12 (improved PGI-I scores: CGAL 88.2%, CGAM 94.8%; mean PGI-I scores: CGAL 2.0, CGAM 1.8).
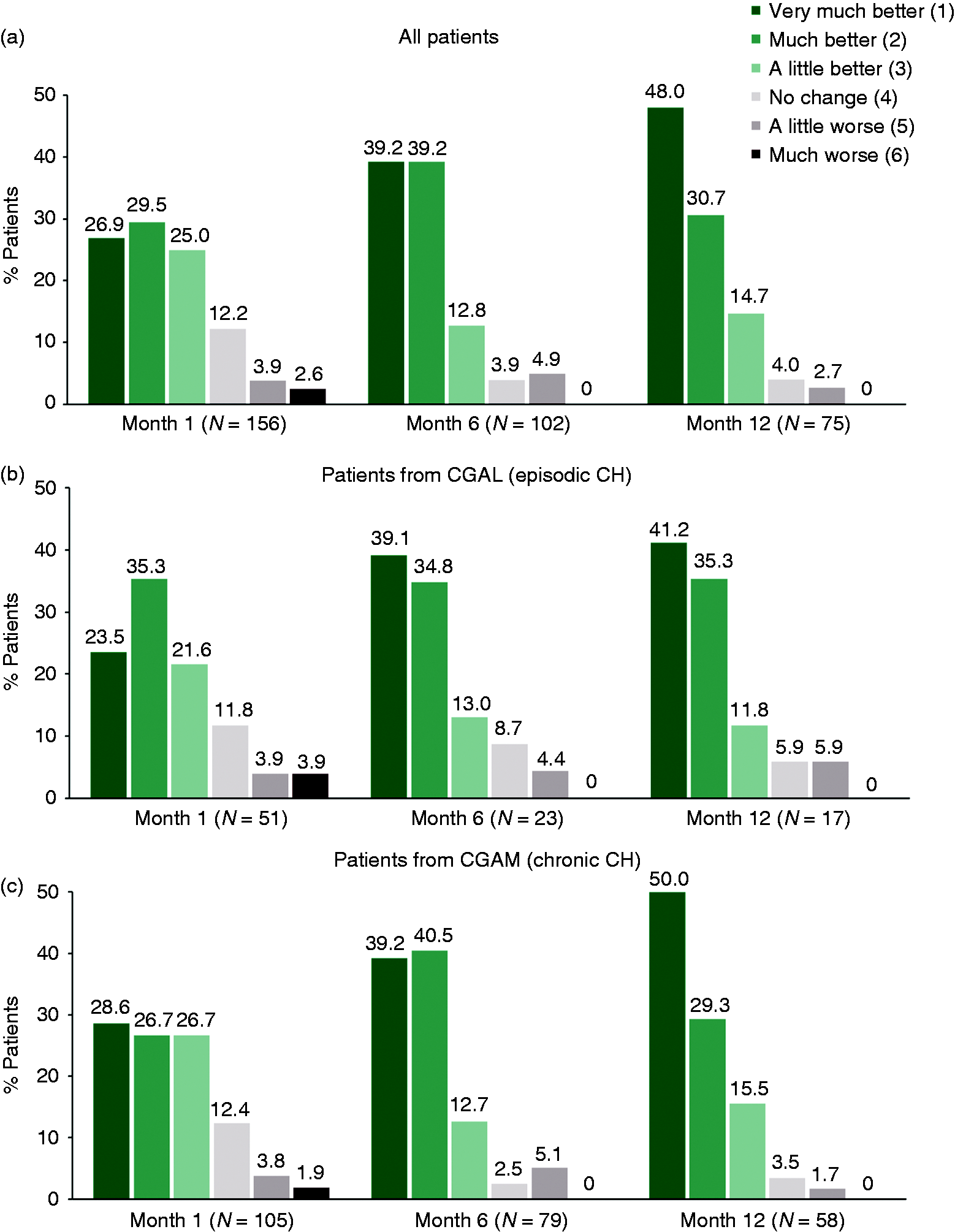
PGI-I over time in (a) all patients, (b) patients with episodic CH, and (c) patients with chronic CH. The PGI-I is a patient assessment of their CH condition since they started taking the medication, based on a patient-rated 7-point scale, where 1 = “very much better,” 2 = “much better,” 3 = “a little better,” 4 = “no change,” 5 = “a little worse,” 6 = “much worse,” and 7 = “very much worse.” Abbreviations: CH = cluster headache; n = number of patients; PGI-I = Patient Global Impression of Improvement.
At CGAR baseline, the majority of patients from each parent study had no problems in the four domains of mobility, self-care, usual activities, and anxiety/depression but reported problems in the domain of pain/discomfort (Table 5). The percentage of patients reporting problems relating to pain/discomfort decreased at Months 1 and 6.
EQ-5D-5L questionnaire categorical analysis.

Abbreviations: EQ-5D-5L = European Quality 5 Dimensions 5-Levels; n = number of patients.
All data reported as n (%).
a“Problems” includes responses of Problem, Moderate Problem, Severe Problem, and Extreme Problem.
bPatients who choose a response of “missing value” are not included in n.
cN = 50.
dN = 104.
In general, health value scores improved at Months 1, 6, and 12, except for EQ-VAS scores at Month 12 for patients from CGAM, where it was relatively unchanged from Month 6 (Figure 3). At Month 1, the mean change in PGI-I was associated with the mean change in Health State Index score (UK) (Spearman’s rank correlation coefficient [r] = −0.30), Health State Index score (US) (r = −0.30), and EQ-VAS (r = −0.33).

(a) Mean EQ-5D-5L Health State Index Score (US), (b) Mean EQ-5D-5L Health State Index Score (UK), and (c) Mean EQ-VAS Current Health Score by parent study. The EQ-5D-5L Health State Index Score is a patient-rated scale with 5 domains (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression); the index ranges between 0 and 1, with the higher score indicating a better health state perceived by the patient. For the EQ-VAS Current Health Score, the EQ-5D-5L score serves as anchors for the patient’s numeric rating on the hash-marked, vertical VAS that ranges from 0 to 100, with the higher score representing the best health the patient can imagine. Abbreviations: EQ-5D-5L = European Quality 5-Dimensions 5-Levels; EQ-VAS = European Quality Visual Analogue Scale; n = number of patients.
Discussion
Patients in CGAR had a mean duration of galcanezumab exposure of between 15 and 16 months, and 26% of patients had at least 24 months of exposure. Thus, it provided additional safety and tolerability data after longer galcanezumab exposures than those in previous galcanezumab studies in migraine (up to one year) and chronic CH (up to 15 months) (6,12). The overall safety profile of galcanezumab in Study CGAR was consistent with that shown in the Phase 2 and 3 studies of galcanezumab for the prevention of episodic or chronic migraine (12) and the Phase 3 CH parent studies, CGAL (5) and CGAM (4). Not only were TEAEs generally mild to moderate in severity, but the rate of discontinuations due to an AE was low (3.1%) and comparable to that observed among patients treated with galcanezumab 240 mg (N = 1350) in pooled migraine studies (3.9%) (12) and lower than that observed in the 52-week extension phase of the galcanezumab CH study (7.7%) (6). In addition, this was the first study to show that if galcanezumab treatment was discontinued then reinitiated days, weeks, or months later, no increases in immunogenicity or hypersensitivity reactions were observed. The efficacy analyses, along with the high retention of patients until study termination and large number of patients with uninterrupted dosing, suggest an overall improvement in health status in patients with CH in response to open-label galcanezumab treatment.
Galcanezumab blocks the activity of calcitonin gene-related peptide (CGRP), which is a potent vasodilator that has been postulated to have a protective role in cardiovascular disease, although clinical studies to date have not shown that CGRP is involved in the homeostasis of BP (13 –16). Additionally, patients with CH may have increased risk for cardiovascular events (17,18). In this study, 13 patients met the criteria for a sustained increase in BP (high BP for at least two consecutive visits); however, the sustained increases were associated with considerable variability, did not persist over time, and were not associated with dosing interruptions or discontinuation. One patient reported an SAE of arterial occlusive disease 141 days after starting galcanezumab and on the same day as the sixth dose. Diagnostic testing showed stenosis of both the right and left common iliac arteries, which was considered by the investigator to not be related to galcanezumab treatment but rather to smoking 30–40 cigarettes/day, and the patient continued in the study. Hypertension was reported as a TEAE in <5% of patients, none of which were severe/serious or led to discontinuation. These findings are consistent with previous CH and migraine galcanezumab studies (4 –6,12).
Although not a primary endpoint of the study, the efficacy of galcanezumab was evaluated on the basis of patients’ assessment of their CH condition using the PGI-I scale. The majority of patients reported their CH condition as very much better or much better beginning one month after initiating galcanezumab treatment, and the percentage increased with longer duration. Although the PGI-I is not specific to CH, in post hoc analyses of the CGAL parent study in patients with episodic CH, PGI-I ratings of feeling better or much better after one month of galcanezumab treatment were associated with other measures of clinical improvement, including reductions in CH attack frequency, duration, and severity (19,20). In the present study, mean changes in PGI-I scores after one month of treatment were associated with mean changes in health value scores. Improvements in the patient-reported domain of pain/discomfort and the overall Health State score, as assessed by the EQ-5D-DL, were also observed and support the findings of the PGI-I assessment. It is also of interest to note that seven patients (four with episodic CH and three with chronic CH) discontinued due to reported lasting remission.
Several limitations warrant consideration. First, potential bias could have been introduced in CGAR due to the open-label nature of the study. Second, other preventive treatments for CH were permitted during the study; however, because the study was primarily a safety study, the potential influence of alternative preventive treatments on efficacy was not examined. Third, the study drug was provided, and it is unknown how this might have influenced the decision to administer a dose of galcanezumab within the conduct of this study. Fourth, there is potential for survivorship bias in which the earlier drop-out of non-responders could have exaggerated the efficacy results; however, the potential for survivorship bias was likely reduced because patients entered CGAR from completed galcanezumab parent studies. For example, at the completion of CGAL, non-responders who knew that they had received galcanezumab would be less likely to enter a new study of the same treatment, especially considering the severity of the CH attacks. This would also be the case in CGAM, in which patients had at least 12 months (and up to 15 months) of monthly dosing. In addition, patients who discontinued either parent study were not eligible to enter CGAR. Considering the points above, if survivorship bias was contributing significantly to the efficacy results, it would most likely be driven by patients from the CGAL placebo arm; however, similar percentages of patients from CGAL and CGAM discontinued due to lack of efficacy (11% and 12%, respectively). It should also be borne in mind that the aim of this study was to assess the long-term safety and tolerability of galcanezumab using a dosing strategy that more closely approximates what may occur in clinical practice, with the effectiveness of galcanezumab being an exploratory objective. Finally, the results of this study may not be fully generalizable to all patients with CH, including, but not limited to patients with unstable or potentially confounding medical condition or at risk of serious or acute cardiovascular events, as they were excluded from the study.
In conclusion, in this open-label, Phase 3b study, galcanezumab 300 mg per month demonstrated a long-term safety profile consistent with that observed in previous Phase 3 studies and durable improvement in patient-reported CH status among patients with episodic and chronic CH.
Clinical implications
CGAR provides longer-term data on galcanezumab 300 mg for the treatment of CH In this open-label safety study, treatment with galcanezumab 300 mg improved the patient-reported CH condition in the majority of patients Galcanezumab 300 mg was shown to have a long-term safety profile that was consistent with that observed in previous Phase 3 CH studies
Supplemental Material
sj-pdf-1-cep-10.1177_03331024221103509 - Supplemental material for Long-term open-label safety study of galcanezumab in patients with episodic or chronic cluster headache
Supplemental material, sj-pdf-1-cep-10.1177_03331024221103509 for Long-term open-label safety study of galcanezumab in patients with episodic or chronic cluster headache by Robert Riesenberg, Charly Gaul, Chad E Stroud, Yan Dong, Mark E Bangs, Richard Wenzel, James M Martinez and Tina Myers Oakes in Cephalalgia
Footnotes
Acknowledgements
Writing support for the development of this manuscript was provided by Cindy C Taylor of Synchrogenix, a Certara company, and funded by Eli Lilly and Company. The authors would like to thank Phebe Kemmer of Eli Lilly and Company for her contributions to the statistical analyses. The study was conducted by Eli Lilly and Company. Some results of this trial were presented at The International Headache Congress – IHS and EHF Joint Congress; Virtual; September 8–12, 2021.
Author contributions
All authors analyzed and interpreted the data and critically revised manuscript drafts for intellectual content. RR and CG also served as principal investigators for the study. YD conducted the statistical analyses.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: CES, TMO, YD, MEB, RW, and JM are employees and minor shareholders of Eli Lilly and Company. RR reports no conflicts of interest outside of participation in this study. CG received honoraria for consulting and lectures within the past three years from Allergan Pharma, Eli Lilly, Novartis Pharma, Hormosan Pharma, Grünenthal, Sanofi-Aventis, Weber & Weber, Lundbeck, Perfood, and Teva. His research is supported by a grant of the German Research Foundation (DFG). He does not hold stocks from any pharmaceutical company. He is honorary secretary of the German Migraine and Headache Society.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The CGAR study was sponsored by Eli Lilly and Company.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.