
Abstract
Background
We present findings from the multicenter, double-blind Phase 3 study, CENTURION. This study was designed to assess the efficacy of and consistency of response to lasmiditan in the acute treatment of migraine across four attacks.
Methods
Patients were randomized 1:1:1 to one of three treatment groups – lasmiditan 200 mg; lasmiditan 100 mg; or a control group that received placebo for three attacks and lasmiditan 50 mg for either the third or fourth attack. The primary endpoints were pain freedom at 2 h (first attack) and pain freedom at 2 h in ≥2/3 attacks. Secondary endpoints included pain relief, sustained pain freedom and disability freedom. Statistical testing used a logistic regression model and graphical methodology to control for multiplicity.
Results
Overall, 1471 patients treated ≥1 migraine attack with the study drug. Both primary endpoints were met for lasmiditan 100 mg and 200 mg (p < 0.001). All gated secondary endpoints were met. The incidence of treatment-emergent adverse events (TEAEs) was highest during the first attack. The most common TEAEs with lasmiditan were dizziness, paresthesia, fatigue, and nausea; these were generally mild or moderate in severity.
Conclusions
These results confirm the early and sustained efficacy of lasmiditan 100 mg and 200 mg and demonstrate consistency of response across multiple attacks.
Introduction
The value of an acute treatment for migraine to a patient depends on its ability to not only relieve symptoms during a migraine attack rapidly and with a sustained effect, but also to deliver efficacy across multiple attacks (i.e. intra-patient consistency of response) (1,2). It is recommended that clinical trials of acute treatments for migraine include assessment of pain and most bothersome symptoms at 2 h, as well as the consistency of response (3,4). Consistency data from placebo-controlled studies have been reported for various triptan drugs, with pain freedom in ≥2 of 3 attacks for 14–42% of patients receiving a triptan compared with 3–13% receiving placebo (5).
Lasmiditan is a selective serotonin 1F (5-HT1F) receptor agonist (ditan), approved by the FDA for the acute treatment of migraine, with or without aura, in adults (6). In two Phase 3 single migraine attack studies, SAMURAI and SPARTAN, lasmiditan demonstrated statistically significant superiority versus placebo in the proportion of patients who were pain free as well as the proportion of patients who were free of their migraine-associated most bothersome symptom (MBS) at 2 h post dose (7,8).
We report findings from the CENTURION study, a Phase 3 placebo-controlled study designed to assess the first attack efficacy of and consistency of response to lasmiditan 100 mg or 200 mg in acute treatment of migraine attacks in individuals with or without aura.
Methods
CENTURION was a multicenter, randomized, placebo-controlled, double-blind modified parallel Phase 3 study conducted in Europe, North America, and Asia. The study was designed to assess the efficacy, including consistency of response, of lasmiditan in the acute treatment of migraine attacks with or without aura.
The study received approval from the relevant Ethics Committees (Supplemental File 1), and patients provided written informed consent for study participation. The study was conducted in accordance with the Declaration of Helsinki.
Patient population
Eligibility criteria (Supplemental File 2) included age ≥18 years (no upper limit), migraine with or without aura fulfilling the International Headache Society (IHS) diagnostic criteria 1.1 or 1.2.1 (9); a history of disabling migraine of at least 1 year; Migraine Disability Assessment Test (MIDAS) (10) score ≥11; migraine onset before the age of 50 years; and 3–8 migraine attacks per month, but <15 headache days per month during the past 3 months. Individuals with known cardiovascular risk factors or disease, with the exception of history of hemorrhagic stroke, were not excluded. Patients on migraine preventive therapies were eligible provided they were stable for 3 months prior to screening.
A subpopulation of patients with an insufficient response to triptans was predefined, based on literature review and input from migraine experts, as patients who met any of the following criteria – an inconsistent response to their most recent triptan, those who were taking a triptan and had a poor/very poor migraine Treatment Optimization Questionnaire (mTOQ-6) score) (11), or had discontinued their most recent triptan for efficacy or tolerability reasons or due to contraindications.
Study design
After a screening visit, qualifying patients attended a second visit at which they were randomized with stratification by country, in a blinded fashion, in a 1:1:1 ratio to one of three treatment groups for four attacks: a) lasmiditan 100 mg; b) lasmiditan 200 mg; or c) a control group, which received placebo for three attacks and lasmiditan 50 mg for either attack 3 or attack 4 (1:1) (Figure 1). To limit differential drop out, patients were not to be informed about the randomization scheme; instead, they were to be informed that no patient would receive placebo for all attacks.

Study design.Abbreviations: LTN, lasmiditan; PBO, placebo; m, monthaVisits at 2 and 3 months applicable where patients had not already treated 4 attacks before these timepoints.bEnd of study visit occurred at ≥7 days after treating the last migraine attack or at 4 months after randomization.
Patients were provided with blinded study drug for treatment of four attacks and were asked to treat consecutive migraine attacks, if possible. For each attack, patients were to treat their migraine within 4 h of onset, providing that the headache severity was at least moderate and not improving, and no other migraine treatment had been taken. Patients were instructed to not take another dose of study drug for ≥48 h after treating an attack with the study drug. If patients were unable to take the study drug and complete the study procedures for an attack, they were permitted to use their usual migraine medication.
The treatment period continued until the four attacks had been treated or until 4 months after randomization, whichever came sooner. Phone visits were conducted after attack 1 or at 1 month after randomization, whichever came sooner, and at 3 months after randomization. A site visit occurred at 2 months after randomization.
For rescue or recurrence, patients were permitted to take their own unexcluded medications beginning at 2 h post dose; patients were not permitted to take triptans, ergotamines, opioids, or barbiturates within 24 h of the study drug.
Training of site staff and patients on the placebo response was provided to ensure neutral interactions between investigative staff and patients and to minimize expectations that could potentially influence study findings.
Efficacy
Study endpoints
Primary endpoints were assessed by treatment group in the following order: a) percentage of patients who were pain free (defined as mild, moderate, or severe headache pain becoming none) at 2 h post dose during the first attack; and b) percentage of patients who were pain free at 2 h post dose in at least two of three attacks (consistency of response). In the event that either active treatment group showed significant differences from placebo for both endpoints a and b, prespecified analyses, controlled to avoid type I error inflation due to multiple comparisons, were performed for key secondary endpoints (gated endpoints). These endpoints were pain relief at 1 and 2 h, pain freedom at 1 h, sustained pain freedom at 24 and 48 h, migraine-related functional disability freedom at 2 h, pain freedom at 2 h in patients with an insufficient response to triptans and consistency of response for pain relief at 2 h.
Additional secondary endpoints included the percentage of patients who were migraine-associated most bothersome symptom (MBS) free at 2 h; who used rescue medication in the 2–24 h period; who reported being “much better” or “very much better”, as measured using the Patient Global Impression of Change (PGIC) (12), at 2 and 24 h; and who were pain free at 2 h and reported recurrence within 24 or 48 h. First attack data were used in all cases.
Assessment of study endpoints
Patients were to record their response to study drug over the 48 h post dose period using an electronic diary (eDiary), with assessments at 0.5, 1, 2, 4, 6, 24, and 48 h.
Patients were asked to record their headache severity before dosing and at the specified post-dose time points using the IHS 4-point headache severity rating scale (9).
Disability was measured by determining the level of interference with normal activities, recorded by the patient at baseline and at the specified post-dose time points, with four response options – “not at all”, “mild interference”, “marked interference”, and “need complete bed rest” – to the question “How much is your migraine interfering with your normal activities?”.
Patients were asked to record their baseline MBS from nausea, phonophobia, or photophobia; freedom from MBS was defined by absence of the chosen symptom at post-baseline time points.
The PGIC, designed as an integrated measure of the effect of treatment on head pain and associated migraine symptoms as well as drug tolerability, was administered at 2 and at 24 h post dose. Patients were asked “How do you feel after taking study medication?” and answered on a 7-point scale (1 = very much better, 7 = very much worse).
Tolerability and safety
For adverse event assessment, patients were asked at each post-dose assessment (using the eDiary), “Do you feel anything unusual since you took the study medication that you have not felt with a migraine before?” If they responded affirmatively, then they were instructed to record relevant information in a paper journal, which was to be reviewed at the next visit. Treatment-emergent adverse events (TEAEs) were defined as new or worsening adverse events during the 48 h after a dose of study drug. Safety assessment also included vital signs and laboratory measures.
Statistical analysis
The study was designed to have >90% power to detect a difference in pain freedom at 2 h during the first attack and pain freedom at 2 h in at least two of three attacks for at least one lasmiditan dose group.
First attack efficacy analyses were conducted using data from the intent-to-treat (ITT) population, which was defined as all randomized patients who used at least one dose of study drug to treat a migraine attack of at least mild pain severity and with any post-dose pain severity assessments at or before 2 h post dose. For analysis sensitivity purposes, analyses were repeated for the modified ITT (mITT) population, which was defined as ITT patients who treated a migraine attack of at least moderate pain severity.
Consistency analyses were conducted using data from the ITT consistency population, which was defined as all patients who experienced at least two successes or two failures during ITT-evaluable attacks (i.e. a treated attack of at least mild pain severity with any post-dose pain severity assessments ≤ 2 h post dose). For the control group, only placebo-treated attacks were considered; for the lasmiditan groups, only the first three ITT-evaluable attacks were considered.
Logistic regression analyses were used for all efficacy analyses. A patient was considered a non-responder if he/she took any other medication for migraine at or before the specific time point. A patient was also considered a non-responder at a specific time point if he/she had an evaluable attack but did not provide the necessary data (e.g. pain severity rating, symptom rating) at that specific time point. A sensitivity analysis using observed case data showed similar results to those from the primary analysis method.
To control for overall type I error, the primary and gated secondary endpoints were tested using a graphical multiple comparisons procedure (13) that preserved overall type I error at a one-sided alpha level of 0.025. The multiple testing scheme is shown in Supplemental Figure 1 and Supplemental Table 1. Initial alphas were assigned to hypotheses comparing pain freedom at 2 h in the first attack for each dose of lasmiditan versus placebo. The tests comparing each dose of lasmiditan with placebo on the coprimary consistency endpoint of pain freedom at 2 h in at least two of three attacks were to be conducted only if the null hypothesis of no pain freedom in the first attack was rejected for the corresponding dose. Testing continued following the prespecified process. If a test was rejected, the alpha for that test was propagated to the other hypotheses as specified in the scheme. Tests of other secondary efficacy endpoints were conducted at a two-sided significance level of 0.05.
The probability of freedom from pain or pain relief over the course of 24 h after dosing was estimated with the use of Kaplan-Meier time-to-event analyses. For each time point, the number of patients eligible to become free from pain or achieve pain relief and the number of patients who reported the first occurrence of the event were used to calculate the probability estimate and 95% confidence intervals.
A pre-specified exploratory analysis (logistic regression model) was performed to compare lasmiditan 50 mg and placebo using the control group data from attacks 3 and 4.
Safety analyses were conducted using data from the safety population, defined as randomized patients who took at least one dose of study drug.
The statistical evaluation was performed using SAS version 9.4 or higher.
Results
The study findings are summarized in Supplemental File 3.
Patients
A total of 1613 patients were randomized (Figure 2), of which 1471 had first attack efficacy data (ITT population) and 1049 provided sufficient data to assess consistency (ITT-consistency population).

Flow of patients through study.Abbreviations: AE, adverse event; W/D, withdrawal.aN for “Completed study” includes 78 patients with no final disposition (e.g., no final onsite visit at clinic completed at time of data cut-off due to Covid-19).
Baseline characteristics were similar across treatment groups (Table 1). Overall, the mean age was 42 years, and the majority were female. More than 70% of patients were in Europe. Across the trial population, the average duration of migraine history was 17 years, the baseline attack frequency 4.9 per month, and the degree of migraine-related disability severe (mean MIDAS score 31.6).
Baseline characteristics.

ITT, intention to treat; IQR, interquartile range; MBS, most bothersome symptom; MIDAS, migraine disability assessment
Europe - Austria, Belgium, Czech Republic, Demark, France, Germany, Hungary, Italy, The Netherlands, The Russian Federation, Spain, Switzerland, United Kingdom; N. America - Mexico, The United States; Asia - China, India
aITT population percentages
bCardiovascular risk factors were age>40 years; systolic blood pressure ≥140 mm Hg and/or medical history of hypertension at baseline; total cholesterol ≥240 mg/dL; and diabetes mellitus
cPatients with multiple symptoms at baseline who did not identify any of them as MBS were considered to have all recorded symptoms as MBS.
Efficacy
First attack
At either dose of lasmiditan, significant differences from placebo were seen for all primary and secondary gated endpoints (p < 0.001 in all cases) (Table 2).
Primary, key secondary and other secondary endpoint findings (ITT population).

CI, confidence interval; h, hours; MBS, most bothersome symptom; NA, not applicable; OR, odds ratio; PGIC, Patient Global Impression of Change; TIR, triptan insufficient responder
‡p<0.01 vs placebo; *p<0.001 vs placebo
aITT population N
bConsistency analyses conducted using data from the ITT consistency or TIR ITT consistency population (patients who experienced at least 2 successes or 2 failures during ITT-evaluable attacks). For the control group, only placebo-treated attacks were considered (N=373); For the lasmiditan 100 mg (N=340) and 200 mg (N=336) dosing groups, only the first 3 ITT-evaluable attacks were considered. First attack data used in all other cases
cPain relief defined as moderate or severe headache pain that became mild or resolved completely, or mild pain that resolved completely
dSustained pain freedom defined as pain freedom at 2 hours
eGated endpoint for lasmiditan 200 mg, secondary endpoint for lasmiditan 100 mg
fNo formal statistical analysis performed
gTotal migraine freedom (defined as no pain or migraine associated symptoms [phonophobia, photophobia, nausea, vomiting]) was a pre-specified exploratory endpoint.
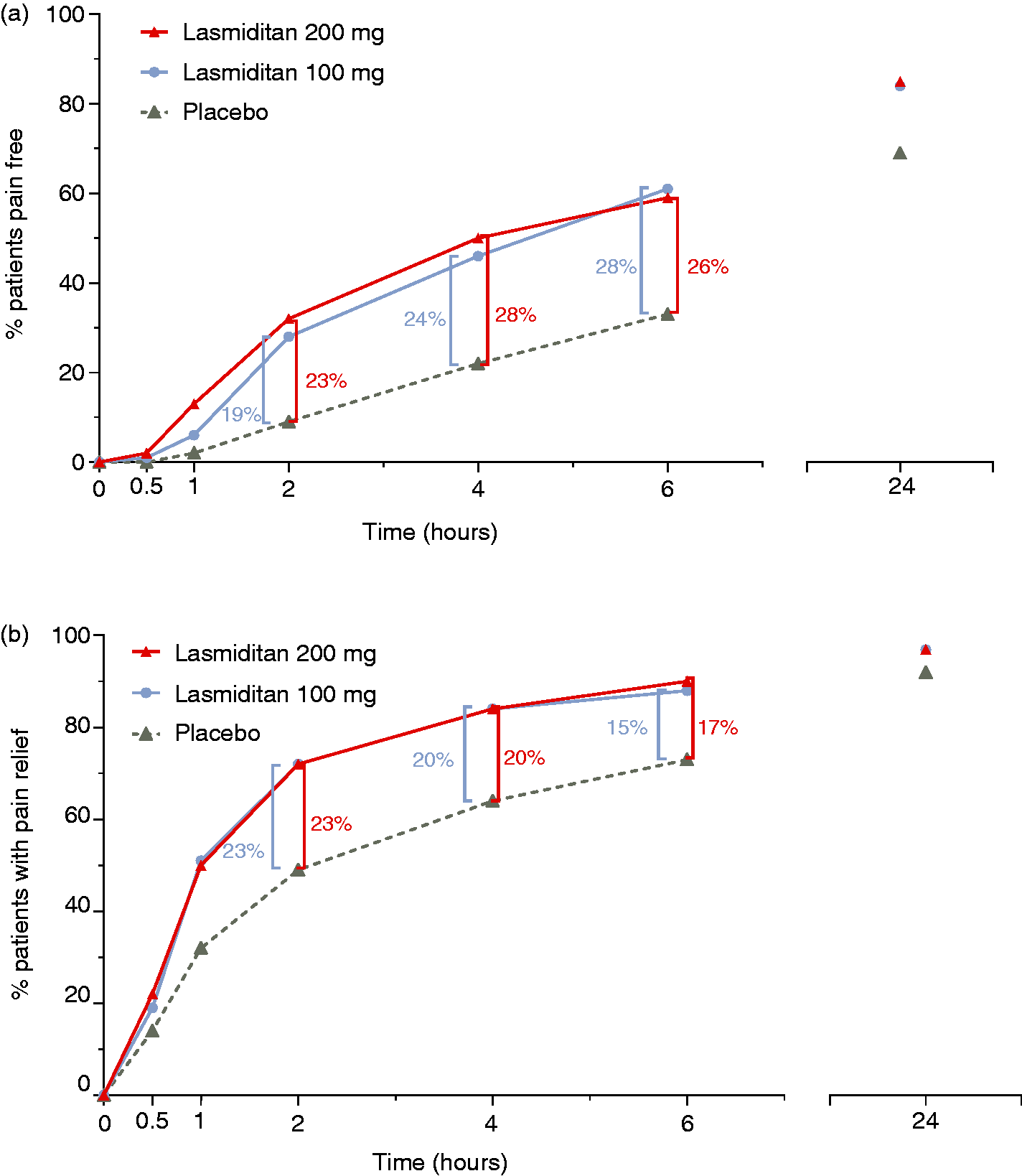
Lasmiditan at either dose was superior to placebo for pain freedom at 2 h (primary endpoint), with therapeutic gains of 17.4% and 20.9% for lasmiditan 100 mg and 200 mg, respectively (Figure 3(a)). There was an early onset of effect (pain freedom at 1 h) for both doses of lasmiditan (Table 2). Time-to-event analysis demonstrated that the effect of lasmiditan on pain freedom was greatest between 4 and 6 h post dose, as evidenced by the separation from placebo at these time points (Figure 4(a)). Both lasmiditan doses had a sustained effect, defined as sustained pain freedom at 24 h or 48 h (Table 2).

Percentage of patients achieving (a) pain freedom and (b) pain relief after first dose.‡ p<0.01 vs placebo; *p<0.001 vs placebo.
Time-to-event analysis (a) pain freedom and (b) pain relief after first dose.
Lasmiditan at either dose was superior to placebo for pain relief at both 1 h and 2 h (Table 2); for lasmiditan 200 mg, separation from placebo started at 30 min (Figure 3b). Time-to-event analysis demonstrated that most benefit versus placebo was seen at 2 h post dose (Figure 4b).
Lasmiditan at either dose was superior to placebo for migraine-related functional disability freedom at 2 h, for feeling very much/much better on the PGIC at 2 h and at 24 h, for MBS freedom at 2 h, for pain recurrence within 24 h or 48 h (in patients who were pain free at 2 h), for use of rescue medication within 24 h post dose, and for total migraine freedom at 2 h (Table 2).
In a predefined subset of patients who had an insufficient response to triptans (efficacy or tolerability issues or contraindications), lasmiditan at either dose was superior to placebo for 2-h pain freedom (Table 2).
Findings from analysis of the mITT population (i.e. excluding attacks of mild severity) were similar to those for the ITT population (p < 0.001 versus placebo for all gated first attack endpoints, data not shown).
Consistency of response
For intra-patient response, lasmiditan at either dose was superior to placebo for pain freedom at 2 h in ≥2 of 3 attacks (primary endpoint), as well as pain relief at 2 h in ≥2 out of 3 attacks (Table 2). Findings from analysis of the mITT population were similar to those for the ITT population (p < 0.001 versus placebo, data not shown).
Lasmiditan at either dose also demonstrated population response, with both doses of lasmiditan showing a consistently greater effect versus placebo across attacks (Figure 5).

Percentage of patients achieving (a) pain freedom and (b) pain relief after first dose across the four attacks.No formal statistical comparisons. Percentages are shown above bars. For Attacks 3 and 4, patients in the control group received either placebo/lasmiditan 50 mg or lasmiditan 50 mg/placebo. Only placebo data are shown on the figure for Attacks 3 and 4 (lasmiditan 50 mg data is presented in Supplemental Figure 1).
Lasmiditan 50 mg
In CENTURION, the control group received lasmiditan 50 mg for either attack 3 or 4 and the pain freedom and pain relief findings are presented by attack in Supplemental Figure 2. Across the two attacks, lasmiditan 50 mg was superior to placebo for pain freedom at 2 h (lasmiditan 50 mg 19.1% vs. placebo 12.0%; p = 0.015) but not pain relief at 2 h (lasmiditan 50 mg 45.8% vs. placebo 39.6%; p = 0.11).
Safety and tolerability
A total of 22 patients (1.5%) reported one or more serious adverse events, with a similar incidence across treatment groups (Table 3). Five participants had a serious adverse event that was considered treatment-emergent.
Adverse event findings (safety population).

TEAE, treatment emergent adverse event.
Patients in the control group received placebo for 3 attacks, and lasmiditan 50 mg for 1 attack (Attack 3 or 4); patients in the lasmiditan 100 mg or 200 mg group received lasmiditan at the specified dose for all 4 attacks.
aOf the serious AEs, 5 were considered treatment emergent: 2 in placebo group (liver disorder, suicidal ideation); 1 in lasmiditan 100 mg group (asthma); 2 in lasmiditan 200 mg group (hemiplegic migraine and serotonin syndrome [met Hunter criteria and Sternbach criteria and considered related to study drug by the investigator]).
bReported in ≥ 2% of patients in any treatment group
cSeverity determined by investigator
During the first attack, a numerically greater proportion of patients in the lasmiditan 100 mg and 200 mg treatment groups (53% and 61%, respectively) reported ≥1 TEAE compared with placebo (22%). TEAEs with lasmiditan were generally neurological in nature and of mild or moderate severity.
The incidence of TEAEs with lasmiditan was highest during the first attack (Table 3). The incidence of TEAEs over the course of the study (up to four attacks) is shown in Supplemental Table 2.
Overall, the changes in laboratory values and vital signs were similar across the treatment groups.
Discussion
In this randomized, placebo-controlled Phase 3 study, lasmiditan was superior to placebo for both primary and all gated secondary endpoints.
During the first attack, lasmiditan at either a 100 mg or 200 mg dose was superior to placebo for pain freedom at 2 h (co-primary endpoint). A significant effect on pain freedom was evident as early as 1 h, and on sustained pain freedom at both 24 and 48 h. At either dose, lasmiditan resulted in pain relief at 1 h, and this commenced as early as 30 min (lasmiditan 200 mg). Lasmiditan at either dose was also superior to placebo for MBS freedom at 2 h, pain recurrence within 24 h or 48 h, and MBS freedom at 2 h. Migraine-related functional disability was improved with lasmiditan and, based on 2- and 24-h PGIC findings, lasmiditan was superior to placebo in terms of the patient’s overall feeling after taking the study medication. In a predefined subset of patients who had an insufficient response to their most recent triptan or had discontinued their most recent triptan for efficacy or tolerability reasons or due to contraindications, lasmiditan at either dose was superior to placebo for 2-h pain freedom.
The CENTURION 2-h pain freedom findings are consistent with those from the Phase 3 SAMURAI and SPARTAN studies (7,8), although there was a lower response rate across all treatment groups in CENTURION; for example, the placebo rate was 8% in CENTURION, and 15% and 21%, respectively, in SAMURAI and SPARTAN. Although these differences could be due to random fluctuation in response rates across trials, efforts were made in the CENTURION trial to neutralize site staff and patient expectations to ensure an accurate assessment of the biological effects of lasmiditan. For example, patients were informed by the sponsor and investigative site staff that they were taking part in a research study and that they should have no preconceived idea how they should feel but instead report truthfully their experience with the study drug. To limit patient expectations of receiving the active study drug, they were not informed of the randomization scheme, only that they would not receive placebo for all attacks.
Across the placebo-controlled Phase 3 trials of lasmiditan, including CENTURION, the therapeutic gains for pain freedom at 2 h post dose were 10.1–17.4% for lasmiditan 100 mg and 16.9–20.9% for lasmiditan 200 mg (7,8). In the current study, a time-to-event analysis demonstrated that both lasmiditan 100 mg and 200 mg exerted late benefit (therapeutic gain at 4 or 6 h: 23–28%, depending on dose and time point).
Patients value a predictable and consistent treatment response across attacks. In this study, lasmiditan at either dose resulted in a consistent intra-patient response, as measured by pain freedom or pain relief at 2 h versus placebo in ≥2 of 3 attacks (Table 2); in the case of pain freedom at 2 h, the response was numerically higher for lasmiditan 200 mg versus 100 mg (24% vs. 14%). Consistency of response at a population level, as measured by the percentage of the population achieving pain freedom or pain relief at 2 h by attack, was also greater with lasmiditan versus placebo (Figure 5). Further support for a response of lasmiditan at a population level is provided by the findings from a 12-month open label safety study in which the proportion of migraine attacks treated with lasmiditan 100 mg or 200 mg that resulted in pain freedom at 2 h post dose was similar across quarters of the study (14).
In CENTURION, no new safety signals were observed. Common TEAEs with lasmiditan were dizziness, paresthesia, fatigue, nausea, vertigo, somnolence, hypoesthesia, muscular weakness, asthenia, and feeling abnormal. These were generally mild or moderate in severity. The incidence of TEAEs was highest during the first attack. The safety and tolerability findings from CENTURION were generally consistent with those from SAMURAI and SPARTAN (15) in terms of type and severity, although the frequency of some TEAEs was higher in CENTURION. Differences in adverse event data collection methodology, the informed consent documents, or other factors (16) in CENTURION may have resulted in a higher reported incidence of TEAEs.
Clinical trials, including CENTURION, have demonstrated that lasmiditan is an efficacious acute treatment for migraine and is also associated with CNS adverse events that are generally mild or moderate in severity. To assess the balance of any positive versus negative effects of treatment, the PGIC asks the question “How do you feel after taking Study Medication?” In CENTURION, significantly more patients reported feeling “much better” or “very much better” in the lasmiditan groups than in the placebo group, both at 2 and 24 h post dose, supporting a positive efficacy-tolerability balance for lasmiditan.
The modified parallel design offers advantages over the random insertion of placebo design typically employed to assess consistency. A similar approach was used to assess the consistency of effect for telcagepant, a molecule previously under development for the acute treatment of migraine (17). With a more typical random-insertion-of-placebo design, patients may know they are likely to receive active study drug for most attacks, leading to high expectations and, potentially, higher response rates. Our modified parallel design allowed for placebo-controlled efficacy data during the first attack and placebo-controlled consistency data across three attacks. In contrast, a random-insertion-of-placebo design generates consistency data without the ability to contextualize the findings based on a comparison to placebo.
There are limitations to this study. The study design included groups randomized to lasmiditan for up to four attacks, but a control group taking placebo for only three attacks. This modified parallel design complicates the interpretation of TEAEs across all attacks, although TEAEs were assessed across treatment groups for each individual attack. The inclusion of a lasmiditan 50 mg treatment for one of the attacks in the control group provides some data for this dose, but the study was not powered for or designed to fully assess the efficacy, consistency, or safety of this dose. In CENTURION, the triptan insufficient responder subset included patients with efficacy or tolerability issues with or contraindications to triptans. There is no universally-accepted definition of a triptan insufficient response. Additional data from this subset will be reported elsewhere.
In conclusion, lasmiditan was superior to placebo for all first attack and response endpoints. Overall safety and tolerability were generally consistent with that observed in previous Phase 3 lasmiditan studies (SAMURAI and SPARTAN) (7,8,15).
Key findings
Lasmiditan is efficacious in the treatment of an acute migraine attack; findings from CENTURION support those from previous Phase 3 lasmiditan studies. Lasmiditan demonstrates consistency of response across multiple migraine attacks. The most common treatment emergent adverse events (TEAEs) with lasmiditan were dizziness, paresthesia, fatigue and nausea; they were generally mild or moderate in severity. TEAEs were most frequently reported during the first treated attack.
Supplemental Material
sj-pdf-1-cep-10.1177_0333102421989232 - Supplemental material for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study
Supplemental material, sj-pdf-1-cep-10.1177_0333102421989232 for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study by Messoud Ashina, Uwe Reuter, Timothy Smith, Judith Krikke-Workel, Suzanne R Klise, Sonja Bragg, Erin G Doty, Sherie A Dowsett, Qun Lin and John H Krege in Cephalalgia
Supplemental Material
sj-pdf-2-cep-10.1177_0333102421989232 - Supplemental material for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study
Supplemental material, sj-pdf-2-cep-10.1177_0333102421989232 for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study by Messoud Ashina, Uwe Reuter, Timothy Smith, Judith Krikke-Workel, Suzanne R Klise, Sonja Bragg, Erin G Doty, Sherie A Dowsett, Qun Lin and John H Krege in Cephalalgia
Supplemental Material
sj-pdf-3-cep-10.1177_0333102421989232 - Supplemental material for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study
Supplemental material, sj-pdf-3-cep-10.1177_0333102421989232 for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study by Messoud Ashina, Uwe Reuter, Timothy Smith, Judith Krikke-Workel, Suzanne R Klise, Sonja Bragg, Erin G Doty, Sherie A Dowsett, Qun Lin and John H Krege in Cephalalgia
Supplemental Material
sj-pdf-4-cep-10.1177_0333102421989232 - Supplemental material for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study
Supplemental material, sj-pdf-4-cep-10.1177_0333102421989232 for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study by Messoud Ashina, Uwe Reuter, Timothy Smith, Judith Krikke-Workel, Suzanne R Klise, Sonja Bragg, Erin G Doty, Sherie A Dowsett, Qun Lin and John H Krege in Cephalalgia
Supplemental Material
sj-pdf-5-cep-10.1177_0333102421989232 - Supplemental material for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study
Supplemental material, sj-pdf-5-cep-10.1177_0333102421989232 for Randomized, controlled trial of lasmiditan over four migraine attacks: Findings from the CENTURION study by Messoud Ashina, Uwe Reuter, Timothy Smith, Judith Krikke-Workel, Suzanne R Klise, Sonja Bragg, Erin G Doty, Sherie A Dowsett, Qun Lin and John H Krege in Cephalalgia
Footnotes
Acknowledgements
We are grateful to all study sites and patients who participated, in Austria, Belgium, China, Czech Republic, Denmark, France, Germany, Hungary, India, Italy, Mexico, The Netherlands, The Russian Federation, Spain, Switzerland, the United Kingdom, and the United States.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JKW, JHK, MA, TS, QL, SRK, SB, EGD, and SAD are full-time employees and minor stockholders at Eli Lilly and Company.
MA is a consultant, speaker or scientific advisor for Allergan, Amgen, Eli Lilly and Company, Lundbeck, Novartis, and Teva, primary investigator for Alder, Allergan, Amgen, Eli Lilly and Company, Lundbeck Novartis and Teva trials; he has no ownership interest and does not own stocks of any pharmaceutical company; he serves as associate editor of Cephalalgia, Headache, and Journal of Headache and Pain; he is the President of the International Headache Society.
TS reports financial relationships with Alder-Lundbeck, Allergan, Amgen, Biohaven, Charleston Labs, Electrocore, Impel, Eli Lilly and Company, Novartis Novo Nordisk, Satsuma, Theranica, United Health Group, and Vorso Technologies.
UR has received speaker fees and honorarium for consulting from Abbvie, Amgen, Allergan, Co-Lucid, Eli Lilly and Company, Lundbeck, Medscape, Novartis, StreaMedUp, and TEVA Pharma; he serves as associated editor of the Journal of Headache and Pain and Frontiers in Neurology and as Board Member of the European Headache Federation.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The CENTURION study was sponsored by Eli Lilly and Company.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.