
Abstract
Hypothesis
To identify genetic factors predisposing to migraine-epilepsy phenotype utilizing a multi-generational family with known linkage to chr12q24.2-q24.3.
Methods
We used single nucleotide polymorphism (SNP) genotyping and next-generation sequencing technologies to perform linkage, haplotype, and variant analyses in an extended Finnish migraine-epilepsy family (n = 120). In addition, we used a large genome-wide association study (GWAS) dataset of migraine and two biobank studies, UK Biobank and FinnGen, to test whether variants within the susceptibility region associate with migraine or epilepsy related phenotypes in a population setting.
Results
The family showed the highest evidence of linkage (LOD 3.42) between rs7966411 and epilepsy. The haplotype shared among 12 out of 13 epilepsy patients in the family covers almost the entire NCOR2 and co-localizes with one of the risk loci of the recent GWAS on migraine. The haplotype harbors nine low-frequency variants with potential regulatory functions. Three of them, in addition to two common variants, show nominal associations with neurological disorders in either UK Biobank or FinnGen.
Conclusion
We provide several independent lines of evidence supporting association between migraine-epilepsy phenotype and NCOR2. Our study suggests that NCOR2 may have a role in both migraine and epilepsy and thus would provide evidence for shared pathophysiology underlying these two diseases.
Keywords
Introduction
Migraine and epilepsy are comorbid, chronic paroxysmal neurologic disorders with episodic manifestations. They share similar triggering factors, clinical and genetic features, and treatment options (1). Patients with one disorder are more likely to have the other compared to the general population. For instance, the risk of migraine in individuals with epilepsy is more than twice as high compared to those without epilepsy (1,2).
To date, in genetic studies of migraine, researchers have mainly identified mutations causing a rare and severe subtype of migraine with aura, hemiplegic migraine. Mutations are located in the CACNA1A (3), ATP1A2 (4) and SCN1A (5) ion channel and transport genes. Mutations in these three genes, in addition to numerous other genes, are also linked to rare monogenic forms of epilepsy. However, mutations in single genes do not seem to be the major cause for migraine with typical aura and migraine without aura, or generalized and focal epilepsies. These common forms of migraine and epilepsy are likely polygenic complex disorders, where both genetic and non-genetic factors account for the disease outcome (1,6,7).
Although migraine and epilepsy are both disorders where dysfunction of neuronal network excitability plays an important role, the detailed pathophysiological mechanism shared by the two disorders is still not fully understood. We hypothesize that identifying genetic susceptibility factors predisposing to both migraine and epilepsy will provide wider understanding of the shared pathophysiology underlying the diseases.
In this study, we aimed to identify such a shared genetic factor, focusing on a susceptibility locus on 12q24.2-q24.3 identified by us earlier in a multi-generational Finnish migraine-epilepsy family (8). To define the migraine-epilepsy susceptibility locus in the studied family, we performed single nucleotide polymorphism (SNP)-based linkage and haplotype analyses followed by family-based variant analyses using next-generation sequencing technologies. We also utilized a large genome-wide association study (GWAS) dataset of migraine and two biobank cohorts to study whether any of the variants, including common variants, within the identified susceptibility region associate with migraine or epilepsy related phenotypes in a population setting.
Material and methods
Patients
The study was based on a large six-generation Finnish migraine-epilepsy family with a total of 120 participants (Supplementary Figure 1, Supplementary Table 1). The studied family is part of the Finnish Migraine Genome Project, which is an extensive family study focusing on clinical and genetic aspects of migraine and its comorbidities, such as epilepsy. Patients with epilepsy in the family were initially identified and recruited through a physician. Since most epilepsy patients in the family also had migraine, the physician contacted all family members and asked for their willingness to participate in the migraine study. All participants gave a written informed consent, and the Ethics Committee of the Hospital District of Helsinki and Uusimaa has approved the study (approval no. 111/13/03/01/2011, 3.8.2011 and HUS/1628/2019 for migraine; and approval no. 424/E7/2002 for epilepsy).
Migraine phenotype data were collected with the validated Finnish Migraine-Specific Questionnaire for Family Studies (9). All diagnoses were based on the criteria of the International Classification of Headache Disorders (ICHD-3 criteria) (10). All epilepsy patients were clinically examined by a neurologist and epilepsy diagnoses were made according to the International League Against Epilepsy criteria (11). Epilepsy diagnoses are presented in more detail in our previous publication (8). Altogether, 110 family members donated a blood sample for genetic studies and genomic DNA was extracted from peripheral blood cells using standard protocols.
The mean participation age of the family members was 31.6 years (SD ± 17.6) and 66.7% of them were women. Of the 120 family members, 69 were diagnosed with migraine, consisting of 43 individuals without aura and 26 with migraine with aura. Furthermore, 23 family members had possible migraine headache, eight individuals had headache without migraine characteristics and 12 individuals reported no headache. Migraine diagnosis was not available for eight individuals, one of them having epilepsy. Altogether, 13 family members were diagnosed with epilepsy (Table 1). Nine out of the 13 epilepsy patients were also diagnosed as having migraine. Three of the epilepsy patients had migraine without aura and six had migraine with aura, of which three were patients with hemiplegic migraine. Clinical features of migraine in the family members with epilepsy are described in more detail in Table 2.
Familial occurrence of migraine and epilepsy in the Finnish migraine-epilepsy family.

Clinical features of migraine in the family members with epilepsy.
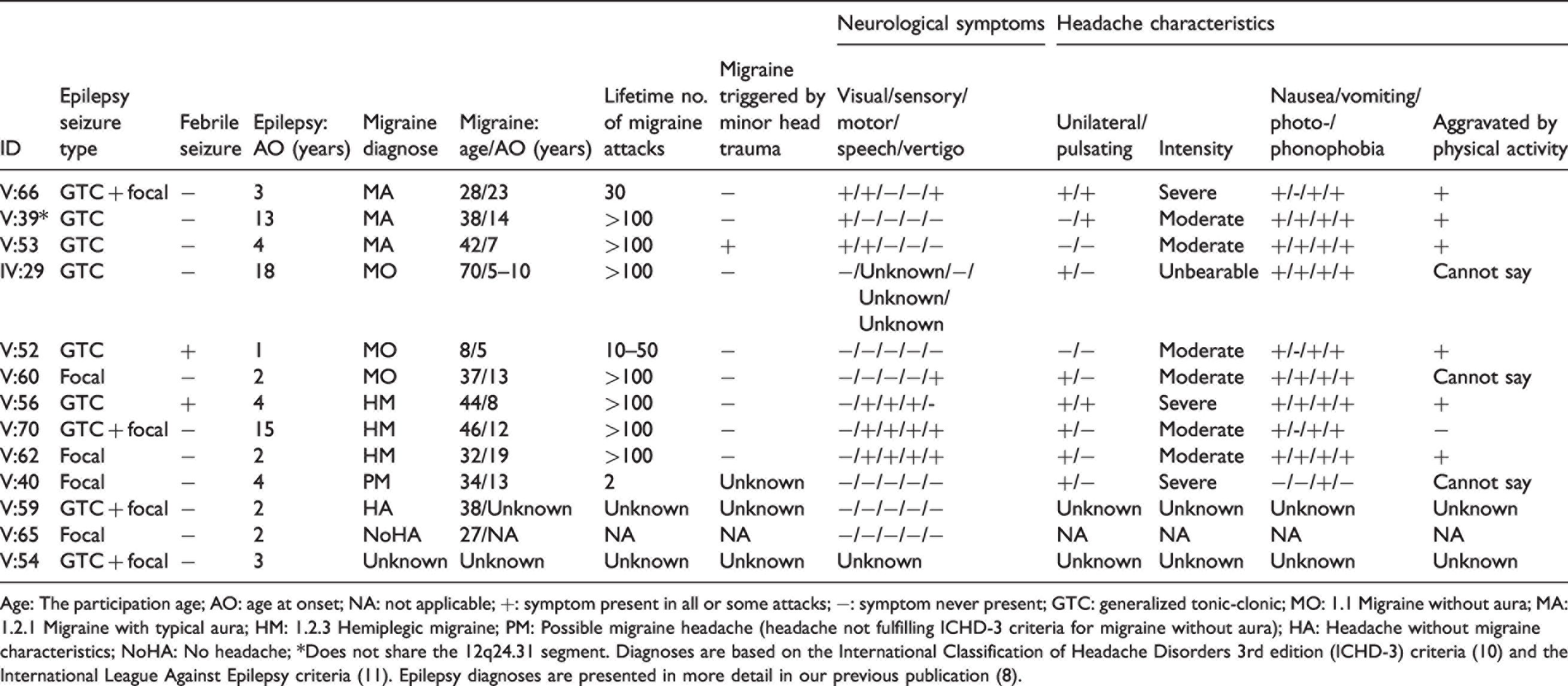
Age: The participation age; AO: age at onset; NA: not applicable; +: symptom present in all or some attacks; −: symptom never present; GTC: generalized tonic-clonic; MO: 1.1 Migraine without aura; MA: 1.2.1 Migraine with typical aura; HM: 1.2.3 Hemiplegic migraine; PM: Possible migraine headache (headache not fulfilling ICHD-3 criteria for migraine without aura); HA: Headache without migraine characteristics; NoHA: No headache; *Does not share the 12q24.31 segment. Diagnoses are based on the International Classification of Headache Disorders 3rd edition (ICHD-3) criteria (10) and the International League Against Epilepsy criteria (11). Epilepsy diagnoses are presented in more detail in our previous publication (8).
Single-nucleotide-polymorphism (SNP) genotyping
Genotyping for linkage analyses was performed for 48 family members at the Technology Centre of the Institute for Molecular Medicine Finland (FIMM), University of Helsinki, using the HumanOmniExpress beadchip (Illumina, San Diego, CA, USA) containing 730,525 markers, of which 35,723 were located on chromosome 12. Genotyped individuals were selected based on their epilepsy and migraine diagnoses and previous information on microsatellite marker alleles on 12q24.2-q24.3. The genotypes were checked for Mendelian incompatibilities using the Pedcheck program (12). All genotyped samples had success rates above 98.9%. In addition to the Illumina HumanOmniExpress beadchip data, we utilized Infinium PsychArray-24 beadchip (Illumina, San Diego, CA, USA) genotype data from 90 family members for haplotype analysis. Altogether, 31 family members were genotyped on both microarrays. The PsychArray genotyping was performed as part of our previous study (6). All genotyped family members are listed in Supplementary Table 1.
Sequencing approaches
To identify potentially causal variants shared by the affected family members, we utilized different sequencing technologies. We selected 48 individuals, including all 13 epilepsy patients, to locus-specific targeted sequencing, eight family members for whole exome sequencing (WES) and four family members to whole genome sequencing (WGS) (Supplementary Table 1). The selection was based on the epilepsy and migraine diagnoses. Sequencing, alignment of sequence reads to reference genome (GRCh37/hg19), quality control and variant detection were completed at the Institute for Molecular Medicine Finland (FIMM), University of Helsinki.
We performed targeted sequencing on a 2.5 Mbp region (chr12:123,751,000-126,250,000; GRCh37) around the linkage peak by using HaloPlex Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA). For WES, we applied SeqCap EZ Human Exome v.2.0 capture kit (Roche NimbleGen, Madison, WI, USA). Both targeted sequencing and WES were performed using the HiSeq platform (Illumina, San Diego, CA, USA) and in house-developed Variant Calling Pipeline (13). WGS data was produced using Chromium Linked-Read sequencing technology (10x Genomics, Pleasanton, CA, USA), NovaSeq (Illumina, San Diego, CA, USA) and 10x Genomics Long Ranger Analysis Pipelines. The average sequencing depth (DP) for targeted sequencing samples was 111, for WES samples 42 and for WGS samples 32. For 90% of the targeted region, the average DP score of all samples was ≥7.
Variant annotation and filtering
To identify associations for risk variants identified in the studied family we used migraine and epilepsy-related results under neurological disease category and we filtered the associations based on the p-value <1 × 10−3. To test whether the susceptibility locus contains other variants associated with these phenotypes, we used same disease category but the associations were filtered based on the suggestive p-value level <1 × 10−5. For the FinnGen PheWeb data we used “VI Diseases of the nervous system” category (ICD-10 Version: 2016) and for the UK Biobank ICD PheWeb data “neurological” category. The UK Biobank PheWeb data was used to identify associations for migraine and epilepsy related medications. In addition to the identified susceptibility locus, we examined genome-wide significant associations (p-value <5 × 10−8) between the NCOR2 gene and all available FinnGen and UK Biobank PheWeb phenotypes.
Statistical methods
To evaluate whether genotyped markers co-segregate with the epilepsy phenotype in the studied family, we performed genome-wide two-point linkage analysis with the recessive and dominant parametric Pseudomarker models (22). Pseudomarker models mimic model-free analyses by using extremely small disease allele frequency and penetrance values (0.00001) without allowing phenocopies (22). Individuals with epilepsy (E: generalized tonic-clonic seizures (GTCS) and/or focal seizures (FS)) were classified as affected and all the other individuals as unknown. Analyses were done with the AUTOGSCAN v2.0 software (23).
We compared family members with the haplotype to family members without the haplotype for 13 clinical variables related to migraine headache and migraine aura. After excluding the spouses, we included all family members with migraine to the analyses. To determine association between the two groups we used Fisher’s Exact test and IBM SPSS Statistics 25.0 software. Missing data and “cannot say” answers were excluded from the analyses. Results with two-sided p-value <0.05 after multiple testing correction (Bonferroni) were considered as evidence of a significant difference between the groups.
Further analysis of the 12q24.31 susceptibility locus using data from the recent migraine GWAS meta-analysis
The 12q24.31 linkage locus was further studied utilizing the largest migraine GWAS meta-analysis dataset of 102,084 affected subjects and 771,257 controls (24). In this study, altogether 10,843,197 markers were tested using the inverse-variance weighted fixed-effects meta-analysis approach. The procedure of the GWAS meta-analysis and the data collections are described in detail in the original publication (24).
We compared the location of the migraine risk locus at 12q24.31 showing evidence of genome-wide significant and suggestive (p-value <1 × 10−5) association with migraine (24) with the haplotype shared by the studied family. We also determined genotypes of the proband IV:29 (mother with epilepsy and migraine) for the migraine GWAS risk variants. In addition, we examined whether the associated locus contained any variants affecting the expression of NCOR2 (nuclear receptor corepressor 2) or SCARB1 (scavenger receptor class B member 1) in neuronal tissues by utilizing the data from the GTEx Portal V8.
Results
Linkage results
The migraine-epilepsy susceptibility locus on chromosome 12q24.2-q24.3 previously identified in this family using microsatellite markers was 13.2 Mbp in size (8). We significantly narrowed the locus using SNP genotyping data (48 individuals) and performing parametric two-point linkage analysis having only the epilepsy patients (n = 12) as affected. The marker rs7966411 (chr12:125,155,819) showed the highest evidence of linkage (logarithm of odds LOD score 3.42, p = 3.6 × 10−5) in the genome-wide linkage analysis (Figure 1). None of the other chromosomal regions achieved LOD score ≥3 (Supplementary Figure 2).

The restricted migraine-epilepsy susceptibility locus on 12q24.2-q24.3. We identified the highest evidence of linkage (logarithm of odds LOD score 3.42, p-value = 3.6 × 10−5) between the marker rs7966411 (chr12:125,155,819; GRCh37) and the epilepsy phenotype (generalized tonic-clonic seizures (GTCS) and/or focal seizures (FS), n = 12). The previous microsatellite susceptibility locus on 12q24.2-q24.3, marked as a rectangle, covered a 13.2 Mbp large region between the markers D12S79 and D12S1659 (chr12:116,160,700-129,316,091; GRCh37) (8).
Shared haplotype at 12q24.31
To further define the locus and to identify the most probable location of the migraine-epilepsy susceptibility variants, we performed haplotype analysis. We defined the haplotype that was shared among the epilepsy patients to be a 450 kbp large region at 12q24.31 (chr12:124,832,606-125,283,766). The region locates between two microsatellite markers D12S1721 and D12S324. Two recombination events centromere-proximal (individual V:65) and centromere-distal (individual V:62) defined the segment (Figure 2). The shared region covers the first 29/47 exons of NCOR2 (nuclear receptor corepressor 2, Ensembl database’s canonical transcript ENST00000405201) and the last 5/13 exons of SCARB1 (scavenger receptor class B member 1, Ensembl database’s canonical transcript ENST00000261693).

Partial pedigree of the Finnish migraine-epilepsy family highlighting the occurrence of epilepsy and migraine in the family and indicating carriers of the 12q24.31 shared haplotype. Sample selection for linkage analysis and targeted sequencing was based on the pedigree.
Twelve of the 13 epilepsy patients shared this haplotype, including two patients (V:56 and V:65) that in the previous linkage study, based on a much sparser microsatellite marker data, were interpreted to be phenocopies (8). In line with the original microsatellite data, there was one epilepsy patient, V:39, that did not share the chromosomal segment segregating with epilepsy in the family. Altogether 38 out of 110 genotyped family members were shown to carry the susceptibility haplotype and rest of the 72 genotyped family members, including all spouses (n = 20), did not share this region (Supplementary Table 1).

Distribution of the migraine load in the studied migraine-epilepsy family.
Headache characteristics and neurological symptoms in the family members with the haplotype compared to the family members without the haplotype.

*All family members with migraine excluding the spouses. Missing data and “cannot say” answers were excluded from the analyses. Questionnaire response rates varied between 81–100% depending on the question.
Susceptibility variants
Despite utilizing different sequencing strategies, we were not able to reveal any rare or common exonic variants with clear functional effect (missense, splicing, stop gain, stop loss, in-frame insertion, in-frame deletion or frameshift) within the shared haplotype at 12q24.31. In addition, no small deletions (50 bp–30 kbp) or large scale structural variants (>50 kbp) within this region were present in the family. However, by combining the different genomic data sources (WES, WGS and targeted sequencing), and applying the filtering criteria described earlier, we identified nine low-frequency (MAF < 0.05) intronic or intergenic variants (Table 4). From the nine variants, rs73227510 was also present in the family members without the shared haplotype (IV:16, V:55, V:21, V:22, V:23, VI:19 and VI:3, Figure 2). Four of the variants (rs75386726, rs11608538, rs11608551 and rs56314649) are in complete linkage disequilibrium (LD) in the European population based on the HaploReg v4.1 database (17).
Details of putative regulatory functions of the shared haplotype variants identified based on the targeted sequencing data on 12q24.31.

Note: Data are derived from the Variant Effect Predictor (VEP, Ensembl Release 75) (15), HaploReg v4.1 (17), RegulomeDB v 2.0 (16) and rVarBase (18); *LD: 1000G Phase 1 population for LD calculation: Europe (HaploReg v4.1). MAF gnomAD v2: minor allele frequency in The Genome Aggregation Database (14).
All the identified nine variants had potential regulatory functions (Table 4). Eight of the variants overlapped with an enhancer in several tissue types, including brain, and two variants (rs11608551 and rs56314649) had RegulomeDB rank of 2b, indicating that the variants are likely to affect binding and are linked to expression of a target gene (16). We did not find any significant eQTLs (expression quantitative trait loci) or sQTLs (splicing quantitative trait loci) for these variants in brain tissue in the GTEx Portal V8. When other tissues were also considered, rs73225443 was a significant eQTL for lincRNA RP11-486O12.2 in artery-tibial tissue and rs73227510 for two other lincRNAs, RP11-83B20.9 and RP11-83B20.8, in testis.
Three of the nine identified variants showed nominal association (p-value < 1 × 10−3) with migraine- or epilepsy-related phenotypes in the UK Biobank data. The variant rs112478330 associated with myoclonus (beta 2.0, p-value 2.1 × 10−4, 95 cases and 395,209 controls) and rs75386726 with other headache syndromes (beta 0.12, p-value 8.5 × 10−4, 7891 cases and 398,780 controls), whereas the variant rs73227510 associated with usage of three different relevant drugs, i.e. fluoxetine (beta 0.0027, p-value 4.2 × 10−5, 6293 item counts, 337,159 samples), topiramate (beta 0.00053, p-value 3.6 × 10−4, 320 item counts, 337,159 samples) and Inderal (beta 0.00035, p-value 8.8 × 10−4, 129 item counts, 337,159 samples). These results are presented in Supplementary Figure 3.
Other nearby variants associating with migraine or epilepsy-related phenotypes
In addition to low-frequency variants, we studied whether any of the variants within the shared haplotype locus associates with neurological traits in the GWAS Catalog, the UK Biobank or the FinnGen cohorts. GWAS Catalog (19) lists altogether 40 association hits and 24 traits. Associated traits include for instance cholesterol levels, lung function and height, but also neurological traits like neuroticism and cognitive performance. In addition to genome-wide significant associations of GWAS Catalog, two common variants showed suggestive associations (p-value <1 × 10−5) with neurological disorders in UK Biobank and FinnGen. One of the variants (rs838503, MAF 0.20 in the Finnish population) showed suggestive association (p-value < 1 × 10−5) with status epilepticus (beta 0.29, p-value 1.6 × 10−6, 767 cases and 243,503 controls) in the FinnGen cohort. The minor allele of the rs838503 variant co-segregates with the shared haplotype in the studied family and the variant is in moderate LD (D´ = 0.78 in the Finnish population) with rs73227510, one of the potentially interesting, less frequent variants discussed earlier (Table 4). The other variant, rs555779825, associated with hemiplegia in the UK Biobank cohort (beta 8.8, p-value 6.3 × 10−7, 1500 cases and 395,209 controls). However, this variant did not co-segregate with the shared haplotype in the studied family.
When performing a full PheWAS analysis for the whole NCOR2 gene, hemiplegia was the sixth most-significantly associated phenotype of all the 1403 electronic health record-based ICD traits available in the UK Biobank data browser. The other five highly associated phenotypes include diaphragmatic hernia, ulceration of intestine, osteoarthrosis, arthropathy associated with neurological disorders, and fracture of tibia and fibula. However, none of these associations was genome-wide significant. Among all 3095 available phenotypes in the FinnGen data, genome-wide significant associations for NCOR2 were observed with coxarthrosis (rs10846644), arthrosis (rs10846653), rheumatological endpoint (rs10846653), gonarthrosis (rs1242997) and atrial fibrillation and flutter (rs6488930). All genome-wide significant associations are presented in Supplementary Table 2.
The migraine risk locus at 12q24.31
One of the 123 migraine risk loci recently reported by the International Headache Genetics Consortium (24) co-localizes with the linked region segregating in the studied family. From these 123 independent loci, 86 were reported for the first time while 37 loci overlapped with the previously reported 48 loci. The lead GWAS SNP rs1271309 (p-value 3.74 × 10−8) is located within the NCOR2 gene, just outside the haplotype shared by the family, and there are eight variants in high LD (r2 > 0.6) with it. The proband IV:29 (mother with epilepsy and migraine) is homozygous for the risk allele for all these nine variants (Supplementary Table 3). The migraine-epilepsy susceptibility region at 12q24.31 is illustrated in detail in Figure 4.
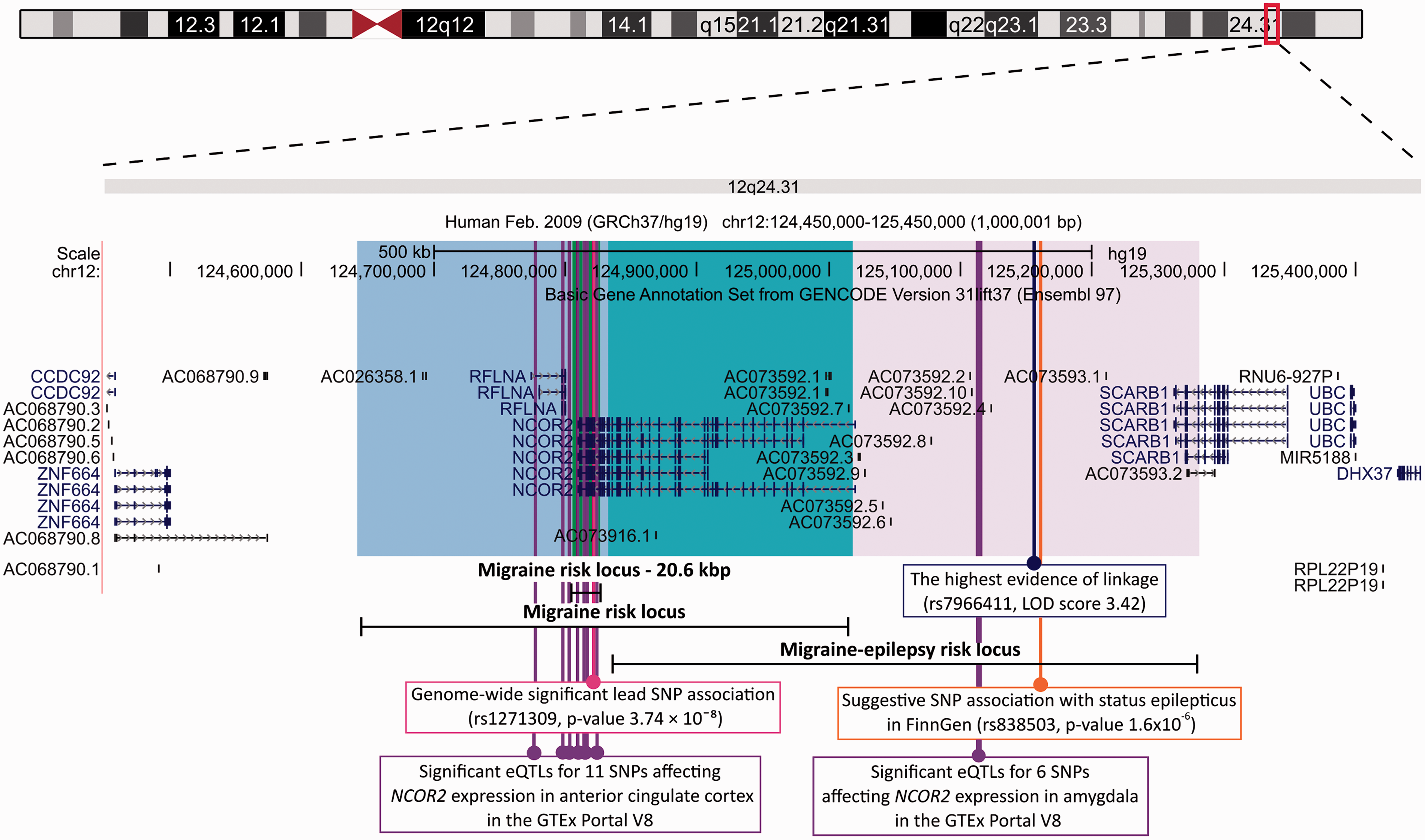
UCSC Genome Browser view (http://genome.ucsc.edu; accessed 06/07/21) (25) of the 12q24.31 migraine-epilepsy susceptibility region. Migraine-epilepsy risk locus shows the position of the shared haplotype on 12q24.31 (chr12:124,832,606-125,283,766; GRCh37) identified in this study. Migraine risk locus was identified in the recent, and so far the largest, genome-wide analysis on migraine (102,084 cases and 77,1257 controls) (24). The risk locus (chr12:124,806,051-124,826,676; GRCh37) covers a 20.6 kbp large region having eight variants in high LD (r2 > 0.6) with the lead variant rs1271309 (p-value 3.74 × 10−8) while the genomic area showing genome-wide significant or suggestive association with migraine (p-value <1 × 10−5) covers a 377 kbp large region on chr12:124,642,694–125,019,242 (GRCh37) (24). The migraine risk locus overlaps with the haplotype segregating in the studied family for a 200 kbp large area on chr12:124,832,606–125,019,242 containing almost the entire NCOR2 gene.
Brain-specific NCOR2 and SCARB1 expression and regulation
Altogether 17 variants in the GTEx Portal V8 were found to be significant eQTLs for the NCOR2 gene in the available brain tissues. Six out of the 17 variants were located on the region shared by the affected family members in our study. All these variants were common (MAF in gnomAD v2 0.4–0.5) and resided in the same European haplotype block based on the HaploReg v4.1 database (17). These variants co-segregated with the identified shared haplotype but were also present in other family members. For all the six variants, the allele present in the shared haplotype leads to lower expression of NCOR2 in amygdala. The remaining 11 variants were located within the recently identified migraine risk locus (24). All the 11 variants affect the expression of NCOR2 in anterior cingulate cortex (Figure 4).
In addition to the eQTLs, there were 13 variants with significant sQTLs for the NCOR2 gene in cerebellum. Three of these variants were located within the shared haplotype. Furthermore, one of the sQTLs variants (rs4765546) was located only 1 kbp from the migraine GWAS meta-analysis lead SNP (rs1271309) (24). However, none of these four variants is present in the family or in the Finnish population based on the gnomAD v2 database (14).
GTEx Portal listed nine variants with significant eQTLs and no significant sQTLs for the SCARB1 gene in the available brain tissues. None of the nine eQTL variants were located within the shared haplotype or migraine risk locus.
Discussion
The present study builds on the previously identified migraine-epilepsy susceptibility locus on 12q24.2–q24.3 segregating in a multi-generational Finnish migraine-epilepsy family (8). We defined a haplotype that was shared among 12 out of 13 epilepsy patients in the family. This 450 kbp large segment on 12q24.31 covers almost the entire NCOR2 gene, the protein of which has an evident brain function. Interestingly, one of the 123 migraine risk loci recently reported by the International Headache Genetics Consortium (24) also co-localizes with this locus. These results suggest that NCOR2 could potentially have a role in both migraine and epilepsy and thus could contribute to the susceptibility of both of these paroxysmal brain diseases.
Co-occurrence of migraine and epilepsy in the family
More than half (69/112) of the family members were diagnosed with migraine and 75% (9/12) of the patients with epilepsy also had migraine. The majority of the epilepsy patients had typical migraine headache characteristics and half of the patients had migraine with aura. This is not surprising considering that epilepsy has been associated in particular with migraine with aura (26). In addition, six family members with epilepsy were under 40 years old when they participated in the study. Thus, their clinical phenotype of migraine may change later in life.
After excluding the spouses, almost all family members with epilepsy (92%) carry the susceptibility haplotype at 12q24.31, but only half of the family members with migraine (49%) carry the region. The family members with migraine who carried the susceptibility region frequently reported ICHD-3 based individual migraine symptoms; for example, unilateral location of headache (56% vs. 25% of the family members without the haplotype) and speech disturbance (36% vs. 8% of the family members without the haplotype). These differences, however, were not statistically significant after multiple testing correction.
Potential regulatory variants within the migraine-epilepsy susceptibility region
The susceptibility haplotype partially harbors two protein coding genes, NCOR2 and SCARB1. However, the variant analysis on the locus did not reveal any potentially damaging exonic variants shared by the affected family members within these genes. Among the shared variants, we highlighted nine low-frequency variants, eight of which were located in the introns of NCOR2 and one in the intergenic region.
Based on data from three functional prediction tools HaploReg v4.1 (17), RegulomeDB v 2.0 (16) and rVarBase (18), all of these highlighted variants had potential regulatory functions. Eight of the variants overlapped with an enhancer in several tissues, including brain, and two variants (rs11608551 and rs56314649) were likely to affect binding and linked to expression of a target gene based on the RegulomeDB rank (16). For seven variants, the potential protein coding target gene was NCOR2.
Furthermore, our PheWAS analyses indicated that, from the identified nine variants, three showed nominal association with migraine- or epilepsy-related phenotypes in the UK Biobank cohort (p-value <1 × 10−3). The associated phenotypes included myoclonus and other headache syndromes, as well as the use of three different drugs; that is, fluoxetine, topiramate and propranolol (Inderal). Topiramate is an anti-epileptic drug, whereas propranolol is a beta blocker medication. Both topiramate and propranolol are effective for migraine prevention (27). Instead, fluoxetine is an antidepressant with potential anticonvulsant properties (28). It can prevent migraine attacks, but there is only limited evidence for its efficacy (27). Even though the small sample sizes of these phenotype subgroups warrant caution for over interpretation, it is intriguing that among the thousands of phenotypes available, the variants of interest showed strongest association with phenotypes related to both migraine and epilepsy.
Common variants within the 12q24.31 shared haplotype associate with migraine- or epilepsy-related phenotypes
In addition to examining these low-frequency variants, we expanded our PheWAS analyses to also cover common variants within the 12q24.31 susceptibility locus. PheWAS analysis highlighted two SNPs associating with migraine or epilepsy related phenotypes. The rs838503 variant showed suggestive association with status epilepticus (p-value 1.6 × 10−6) in the FinnGen cohort. The variant co-segregates with the shared haplotype in the studied family and is in moderate LD with rs73227510, one of the potential regulatory and low-frequency variants identified in this study. Interestingly, rs73227510 is the variant that associated with fluoxetine, topiramate and propranolol use in the UK Biobank cohort as discussed earlier. Another variant, rs555779825, showed suggestive association with hemiplegia in the UK Biobank. In fact, when the whole NCOR2 gene was considered, hemiplegia was the sixth highly associated phenotype of all the 1403 electronic health record-based ICD traits available in the UK Biobank data browser. Respectively, when the whole NCOR2 gene and all available 3095 phenotypes in the FinnGen data were taken into consideration, altogether four SNPs showed genome-wide significant associations with either coxarthrosis, arthrosis, rheumatological endpoint, gonarthrosis or atrial fibrillation and flutter. Interestingly, NCOR2 has previously been linked to osteoarthritis in epigenetic analyses (29) and both migraine and epilepsy are comorbid with autoimmune diseases such as rheumatoid arthritis, and heart diseases such as stroke (30,31).
Migraine risk locus co-localizes with the shared haplotype
Further support for the identified migraine-epilepsy susceptibility locus on 12q24.31 was obtained from the results of the recent, and thus far, the largest genome-wide analysis on migraine (24). One of the identified 123 migraine risk loci co-localizes with the 12q24.31 shared haplotype, with the lead SNP rs1271309 locating within the NCOR2 gene. The genomic area showing genome-wide significant or suggestive association with migraine overlaps with the haplotype segregating in the studied family for a 200 kbp large area containing almost the entire NCOR2 gene. The proband IV:29 is homozygous for the migraine risk allele for the lead SNP and its high LD variants.
Regarding epilepsy, there are 16 genome-wide significant loci identified for common epilepsy, highlighting a potential gene regulatory function in the brain and including candidate genes of known epilepsy genes, as well as targets of antiepileptic drugs (32). However, none of the identified 16 loci were located on the 12q24.31 migraine-epilepsy susceptibility locus. Based on the summary statistics available from the study, we detected that the highest evidence of association for the region is seen between the variant rs701078 and generalized epilepsy (p-value 7.1 × 10−5). The variant rs701078 is a common (MAF in Finnish 0.21) intergenic variant located 11.4 kbp from the lead linkage variant (rs7966411). It is possible that this locus would also reach the level of genome-wide significance in an epilepsy GWAS with a larger sample size.
NCOR2 is a potential candidate gene for migraine and epilepsy
The results of our study support the role of NCOR2 as a more likely migraine and epilepsy gene than SCARB1. The shared haplotype covers almost the entire NCOR2 gene and its promoter region, but only the 3’ end of the SCARB1. In addition, all the potential regulatory variants identified in this study point to NCOR2. The data from the GTEx project V8 also provides more support for the involvement of the NCOR2 gene. Altogether, 17 variants in the GTEx Portal were found to be significant eQTLs for NCOR2 in the available brain tissues. Six out of the 17 variants were located on the region shared by the affected family members in our study. For all the six variants, the allele segregating with the shared haplotype leads to lower expression of the NCOR2 in the amygdala. The remaining 11 variants were located within the migraine risk locus. They all affect the expression of NCOR2 in the anterior cingulate cortex. Interestingly, both brain regions, the amygdala and the anterior cingulate cortex, have been associated with epileptic headache, and more generally, pain processing (33). On the contrary, none of the nine eQTL variants for the SCARB1 gene in the available brain tissues were located within the 12q24.31 migraine-epilepsy susceptibility region.
NCOR2 is an essential transcriptional co-repressor expressed in almost all tissues including the brain, where it participates in forebrain development and neuronal survival (34), whereas the SCARB1 gene encodes a plasma membrane receptor and mediates uptake of cholesterol (35). Thus, based on the literature, both genes could be regarded as potential candidate genes for migraine and epilepsy. However, NCOR2 has a more evident brain function compared to SCARB1 and its mutations have been suggested to cause an atypical form of Rett syndrome (36). Rett syndrome is a rare and severe disorder with different types of symptoms including seizures, motor disturbances, autistic-like behavior, loss of speech, breathing issues and scoliosis typically caused by mutations in the MECP2 gene (37). Studies in mice have shown that the main brain dysfunction in Rett syndrome is due to a loss of the MeCP2-bridge between the DNA and NCOR2-HDAC3 (HDAC3, histone deacetylase 3) co-repressors complex. NCOR2-HDAC3 complex cannot bind to chromatin, resulting in increased transcription of target genes (38,39).
Since NCOR2 has been shown to be intolerant for loss-of-function variants and its genetic deletion leads to embryonic lethality (34), it is not surprising that such variants were not identified in the studied family. Based on the gnomAD v2 data (14) only 10% of the expected loss-of-function variants (o/e score 0.1, 90% confidence interval: 0.06–0.17) were observed for the NCOR2 gene. It is thus possible that damaging NCOR2 variants cause more severe multi-organ symptoms, for instance Rett syndrome-like phenotypes (36). We hypothesize that a regulatory variant affecting the expression of NCOR2 in the brain is a possible cause for the increased susceptibility for migraine-epilepsy attacks seen in this family.
Strengths and limitations
The main strength of this study is the comprehensive and high-quality clinical and genetic data available. We have applied various study designs and made extensive use of existing research data to be able to significantly narrow the migraine-epilepsy susceptibility locus segregating in the studied family.
All epilepsy phenotypes are based on interviews and clinical examinations. Migraine clinical data has been collected with a combination of individual interviews and with the extensive, validated Finnish Migraine-Specific Questionnaire for Family Studies (9). Although questionnaire-based studies provide advantages, it is also important to recognize that the self-reported data has its own limitations and is not equivalent to data derived from clinical interviews. However, the questionnaire is very thorough and includes, in addition to multiple choice questions, parts where participants describe in their own words headache characteristics and draw details of their visual or motor aura. All epilepsy phenotypes are based on interviews and clinical examinations.
To make sure that we did not miss any rare co-segregating variant in the region, we complemented the targeted sequencing data with WGS and WES data. Similarly, WGS and WES data were used to confirm that the nearby region outside the shared haplotype did not contain any rare exonic and potentially damaging variants. We also excluded chromosomal rearrangements as the possible genetic cause. Thus, we can be confident that if an obvious variant were present in the region, our methods would have detected it.
Article highlights
This study supports the hypothesis that migraine and epilepsy share genetic susceptibility factors. Our results provide several independent lines of evidence connecting migraine-epilepsy phenotype and the NCOR2 gene, one of the candidate genes identified also in the latest genome-wide analysis on migraine (24). NCOR2 plays an essential regulatory role during brain development, making it a strong functional candidate gene for neurological disorders and worthy of further functional studies.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-pdf-1-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Supplemental Material
sj-pdf-2-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-pdf-2-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Supplemental Material
sj-pdf-3-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-pdf-3-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Supplemental Material
sj-xlsx-4-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-xlsx-4-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Supplemental Material
sj-pdf-5-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-pdf-5-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Supplemental Material
sj-pdf-6-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-pdf-6-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Supplemental Material
sj-pdf-7-cep-10.1177_03331024211068065 - Supplemental material for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype
Supplemental material, sj-pdf-7-cep-10.1177_03331024211068065 for NCOR2 is a novel candidate gene for migraine-epilepsy phenotype by Marjo Eveliina Nuottamo, Paavo Häppölä, Ville Artto, Heidi Hautakangas, Matti Pirinen, Tero Hiekkalinna, Pekka Ellonen, Maija Lepistö, Eija Hämäläinen, International Headache Genetics Consortium (IHGC), FinnGenConsortiumAuli Siren, Anna-Elina Lehesjoki, Mikko Kallela, Aarno Palotie, Mari Anneli Kaunisto and Maija Wessman in Cephalalgia
Footnotes
Acknowledgements
We gratefully acknowledge the UK Biobank study and all the family members who have participated in the study.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: VA has served on Advisory Boards for Allergan, TEVA, Lilly and Lundbeck, has received funding for travel and/or speaker honoraria from TEVA, Novartis, and Sanofi; has received compensation for producing educational material from Lilly, TEVA and Novartis. MK has served on Advisory Boards for MSD, Allergan, TEVA, Lilly and Lundbeck, has received funding for travel and/or speaker honoraria from MSD, Allergan, TEVA, Novartis, and Genzyme; has received compensation for producing educational material from TEVA and Allergan; has received research support from Helsinki University Central Hospital; and holds stock/stock options and/or has received Board of Directors compensation from Helsinki Headache Center. AP is a member of AstraZeneca Genomics Advisory Board; and is the Scientific Director of the public-private partnership project FinnGen, funded by 12 international pharmaceutical companies and Business Finland. Part of MAK’s salary is covered by the public-private partnership research project FinnGen.
Other authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Folkhälsan Research Foundation, Finland (to MW); The Academy of Finland [grant number 137950 (to MW)]; Academy of Finland Centre of Excellence in Complex Disease Genetics [grant numbers 312074, 336824 (to AP)]; Medicinska Understödsföreningen Liv och Hälsa rf (to MW); the Sigrid Juselius Foundation, Finland (to AP) and Helsinki University Central Hospital (to MK). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.