
Abstract
Background
Knowledge of exact signalling events during migraine attacks is lacking. Various substances are known to trigger migraine attacks in patients and calcitonin gene-related peptide antagonising drugs are effective against migraine pain. Here, we investigated the signalling pathways involved in three different mouse models of provoked migraine and relate them to calcitonin gene-related peptide and other migraine-relevant targets.
Methods
In vivo mouse models of glyceryl trinitrate-, cilostazol- and levcromakalim-induced migraine were applied utilising tactile sensitivity to von Frey filaments as measuring readout. Signalling pathways involved in the three models were dissected by use of specific knockout mice and chemical inhibitors. In vivo results were supported by ex vivo wire myograph experiments measuring arterial dilatory responses and ex vivo calcitonin gene-related peptide release from trigeminal ganglion and trigeminal nucleus caudalis from mice.
Results
Glyceryl trinitrate-induced hypersensitivity was dependent on both prostaglandins and transient receptor potential cation channel, subfamily A, member 1, whereas cilostazol- and levcromakalim-induced hypersensitivity were independent of both. All three migraine triggers activated calcitonin gene-related peptide signalling, as both receptor antagonism and antibody neutralisation of calcitonin gene-related peptide were effective inhibitors of hypersensitivity in all three models. Stimulation of trigeminal ganglia and brain stem tissue samples with cilostazol and levcromakalim did not result in release of calcitonin gene-related peptide, and vasodilation following levcromakalim stimulation was independent of CGRP receptor antagonism.
Conclusion
The mouse models of glyceryl trinitrate-, cilostazol- and levcromakalim- induced migraine all involve calcitonin gene-related peptide signalling in a complex interplay between different cell/tissue types. These models are useful in the study of migraine mechanisms.
Introduction
Migraine treatment has been revolutionized by antibodies and small molecules targeting calcitonin gene-related peptide (CGRP) or its receptor (1). These CGRP antagonizing drugs were developed following basic and clinical research establishing CGRP as a migraine relevant target (2,3) without distinct knowledge about its site and mechanism of action in migraine (4,5). Moreover, not all migraine patients respond satisfactorily (6), suggesting that not all migraines are mediated through CGRP.
Besides CGRP (2), various other trigger compounds such as nitric oxide (NO) donor glyceryl trinitrate (GTN), phosphodiesterase 3 (PDE3) inhibitor cilostazol and ATP-sensitive potassium (KATP) channel opener levcromakalim are triggers of headache in healthy subjects and of migraine attacks in migraine patients following systemic administration (7–11). The latter three substances are different by nature and have distinct pharmacodynamic actions. GTN works via the soluble guanylyl cyclase (sCG) and the cyclic guanosine monophosphate (cGMP) pathway (12) and also generates reactive oxygen and nitrogen species that activate different types of transient receptor potential (TRP) channels (13–15). Cilostazol acts intracellularly by inhibiting PDE3, which breaks down cyclic adenosine monophosphate (cAMP). The net effect of cilostazol is an increase in intracellular cAMP (16). CGRP also increases intracellular cAMP upon binding to its receptor (3). In contrast, levcromakalim acts by direct opening of KATP channels causing K+ efflux, hyperpolarization of the membrane and decreased intracellular Ca2+ (17). All these substances are potent vasodilators and trigger migraine in patients. Both the increase in cGMP mediated by GTN, and in cAMP by cilostazol, facilitate opening of KATP channels (17), which in turn causes relaxation of vascular smooth muscle cells. Therefore, GTN, cilostazol and levcromakalim share at least one mechanism that results in arterial dilation. Cilostazol and levcromakalim act downstream from the CGRP receptor (3,17).
In the present study we investigated if the pathway is also shared in terms of the migraine-inducing effect of the three substances. This was done by repeatedly injecting GTN, cilostazol and levcromakalim to wild type (WT) and specific knockout (KO) mice and by chemical inhibition of different targets. Our findings support the involvement of both shared and independent pathways and reveal dependency of CGRP and its receptor of all three migraine triggers. This is surprising because CGRP in single cells works upstream from cilostazol and levcromakalim.
Materials and Methods
Experimental animals
Experiments were performed under license number 2017-15-0201-01358 from the Danish Animal Experiments Inspectorate and in accordance with the European Community guide for the care and use of animals (2010/63/UE). A total of 617 mice were used to complete the study of which 10 were excluded during in vivo experiments for reasons provided in Table 1. Mice were weighed on every test day for dosing purposes and general health assessment. Subjects were adult male and female mice 8-14 weeks of age on C57Bl/6J background: Wild type mice were C57Bl/6JBomTac (Taconic, Denmark), Trpa1 knockouts (Trpa1tm1/Dpc (18) in-house breeding derived from breeders donated by Peter Reeh, Friedrich-Alexander-University of Erlangen-Nürnberg), Ramp1 knockouts and WT littermates (B6.129S2(Cg)-Ramp1tm1.1Tsuj/WkinJ (19) in-house breeding derived from Jax stock #031560, US). Mice were group-housed in a climate-controlled room under a 12 h light/dark cycle with lights on at 7 AM. Food and water were freely available, and cages enriched with multiple shelters and two types of nesting material as previously described in detail (20).
List of mice excluded during in vivo experiments. All reasons were completely random.

Mouse models
Three different mouse models of provoked migraine were applied. The GTN model is commonly used and thoroughly validated for its relevance to migraine (20–22). The levcromakalim (23) and cilostazol models are novel; the latter presented here for the first time, but previously described in rats (24). Hypersensitivity to tactile stimulation was induced by repeatedly injecting the migraine trigger every other day for a total of 5 times, resulting in test days being day 1, day 3, day 5, day 7 and day 9. On every test day mice were evaluated prior to injection of test substances (basal sensitivity) and at relevant timepoints after injection, which is 2 h for GTN and levcromakalim and 6 h for cilostazol. Antagonising drugs were given 15 minutes prior to the migraine-provoking substance apart from antibodies that were injected only once, 24 h before the first test day. As a surrogate measure of pain (25–27), tactile sensitivity to stimulation with von Frey monofilaments (Ugo Basile) within periorbital or hind paw nociceptive circuits was measured using the up-down paradigm (28,29). Calculation of 50% withdrawal thresholds was done using the free online calculator at https://bioapps.shinyapps.io/von_frey_app/ with application of exact inter-filament steps and target force as input variables (30), starting stimulation with the 0.4 g filament for plantar testing and 0.16 g for cephalic testing. Plantar testing was performed with the mouse placed in clear plexiglas chambers on a mesh floor (IITC Life Science). Mice were placed in the chambers for acclimatization 30–45 minutes prior to testing (27). For periorbital testing, a round plastic cup of 5.8 cm height and 8 cm diameter was placed with the bottom surface facing upwards within each individual plexiglass cage serving as a platform for the mouse to rest on and enabled the experimenter to access the periorbital area with von Frey filaments from above. Mice were placed on the platform 1 h prior to testing and two times prior to the first test (27). Periorbital testing was not performed on days 3 and 7 to avoid sensitizing the mice to the test. All testing was performed between 8 am and 4 pm by an experimenter blinded to treatment groups.
Motor function
To demonstrate that the von Frey test was not biased by impaired motor function or sedation as a potential side effect of drug combinations, general motor function was assessed using a rotarod test paradigm following injection of selected combinations of compounds. Briefly, mice were trained to stay 2 min on a rotarod (LE8200 Panlab, Harvard Apparatus) at 15 rpm prior to drug challenges. Following one-time injections, mice were tested at same time points as for von Frey testing. Each mouse was given three attempts on the rotarod and the longest stay (max 2 min) was recorded. Midazolam, 1 mg/kg IP given 10 min prior to testing was used as positive control. The same 24 mice were used to test the three different drug combinations with 4-5 days washout between and their ability to perform the test assessed on every test day prior to drug administration. Mice were randomised on every test day to receive either vehicle, active drug or midazolam
Ex vivo vasoactivity
Mice were anaesthetized in gas (70% CO2 and 30% O2) and decapitated. The brain was carefully released from the skull and immersed in cold oxygenated Ca2+ free Na+Krebs buffer (in mM: 119 NaCl, 4.6 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 15 NaHCO3, 5.5 glucose and 0.03 EDTA; pH 7.4). The basilar artery was isolated and divided into two segments of 1–2 mm. Segments were mounted on 25 µm diameter wires in a Mulvany-Halpern wire myograph (Danish Myo Technology) with 37°C Na+ Krebs buffer (in mM: 119 NaCl, 4.6 KCl, 1.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 15 NaHCO3 and 5.5 glucose; pH 7.4). Following 15 min of equilibration, the vessels were stretched to reach a pretension of 1–2 mN/mm. Initially, K+ Krebs (similar to Na+Krebs, except 55.4 mM NaCl is exchanged for KCl, making it 60 mM K+) was added to examine the contractile potential of vessels. Half of the segments were treated with 1 µM olcegepant (MedChemExpress). After 30 min of stabilization, all segments were exposed to 0.3 µM U46619 (Tocris, bio-techne) to induce a stable precontraction. Vasorelaxation was studied as a cumulative concentration-response curve to levcromakalim (0.01–10 µM) (Tocris, bio-techne). Following levcromakalim-induced vasodilation, a cumulative concentration-response curve to glibenclamide (0.001–1 µM) (Tocris, bio-techne) was performed. Myograph responses were collected in LabChart™ (ADInstruments). Percent of precontraction was calculated as
Ex vivo CGRP release
The ability of super cinnamaldehyde, cilostazol, forskolin, 8-Br-cAMP and levcromakalim to stimulate CGRP release from the trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC) was tested in an ex vivo assay as previously described for rats (31). Wild type and Trpa1 KO mice aged 8 weeks were euthanized by decapitation following gas anaesthesia (70% CO2 and 30% O2). Tissue samples were washed in synthetic interstitial fluid (SIF, in mM: 108 NaCl, 3.48 KCl, 3.5 MgSO4, 26 NaHCO3, 11.7 NaH2PO4, 1.5 CaCl2, 9.6 NaC6H11O7, 5.5 glucose, 7.6 sucrose; pH 7.4) for 30 min at room temperature, replacing SIF every 5 min. Subsequently, tissues were placed in a 37°C incubator and washed five times with 250 µl of SIF; the first four washes of five min and the last of 10 min. Following the final wash, 200 µl of the SIF was sampled to express basal level of CGRP release from the tissue. Next, tissues were incubated for 10 min with active compound or vehicle and 200 µl samples were obtained. Finally, all tissues were incubated with 10 µM capsaicin as a positive control for the ability of the tissue to release CGRP. CGRP levels were then measured with a commercially available rat or human ELISA kit (Bertin Bioreagent) using the standard protocol supplied by the manufacturer.
Immunofluorescence
Mice were deeply anaesthetized with sodium pentobarbital and transcardially perfused with PBS and subsequently 25 ml PFA 4% (Hospital Pharmacy). The trigeminal ganglia were dissected and stored in PBS. Serial sections, 3–5 μm thick, were cut in transverse planes and placed on silanized slides and stored at 4°C. For immunofluorescence microscopy, sections were deparaffinized and rehydrated in xylene followed by a series of graded alcohol in accordance with established procedures, treated with TEG buffer before blocking with Dako REAL™ Antibody Diluent (Agilent) and left in primary antibodies; mouse monoclonal acetylated alpha tubulin 1:2000 (Sigma-Aldrich, T7451) and rabbit polyclonal TRPA1 1:200 (Alamone labs, ACC-037) overnight at 4°C in a humidified chamber. After PBS washing steps, slides were incubated in corresponding Alexa Flour® secondary antibodies (Invitrogen, A-10037 and A-2106) 1:600 for 45 min at room temperature. Cell nuclei were labelled by DAPI staining. Coverslips were mounted with either Dako fluorescent mounting medium or with PBS containing 90% glycerol, and 2% N-propyl-gallate and subjected to epifluorescence microscopy. Images were captured on a fully motorized Olympus BX63 upright microscope with an Olympus DP72 color, 12.8-megapixel, 4.140 × 3.096-resolution camera and with a fully motorized and automated Olympus IX83 Inverted microscope with a Hamamatsu ORCA-Flash 4.0 camera (C11440-22CU). The software used was Olympus CellSens dimension, which was able to do deconvolution on captured z stacks, and images were processed for publication using Image J and Adobe Photoshop CS6.
Test compounds
Substances used in vivo were administrated intraperitoneally (IP) or orally by gavage (PO) in a volume of 10 ml/kg and vehicle was identical to that of the active compound. Details on in vivo compounds and dosing are provided in Table 2. For ex vivo experiments, substances were dissolved in dimethyl sulfoxide (DMSO) and further diluted as indicated. Ex vivo cilostazol, super cinnamaldehyde (TRPA1 agonist), forskolin (adenylate cyclase stimulator), 8-Br-cAMP (cAMP analogue) and capsaicin (TRPV1 agonist) were from Sigma-Aldrich and U46619 from Tocris, Bio-Techne.
Compounds used in vivo. CMC = carboxymethylcellulose. Cilostazol tablets of 100 mg were crushed to a fine powder and suspended in the vehicle solution. HC030031 was also given as a suspension.

Statistical analyses
Group sizes for in vivo experiments were based on our own previous work with the mouse model, where adequate power for detection of an intermediate effect of antagonising drugs was reached with group sizes of 10–12 in repeated measure two-way ANOVA designs with 3–4 treatment groups and 5 days of multiple comparisons (23,27,32). Thus, no formal power calculation was performed as the many variables (33) in these makes it complicated to arrive at the relevant real-life group size. Smaller groups were used in initial dose finding experiments as these were replicated in the experiments confirming cephalic sensitivity and with addition of antagonists. Mice were randomised to test groups by draw but counterbalanced in relation to baseline sensitivity (provided as supplementary data for all figures) and home cage to avoid confounding by these two factors. Mechanical sensitivity data were square root (SQRT) transformed for improved Gaussian distribution. Thus, data are presented as mean ± SEM of SQRT [50% withdrawal threshold in grams]. Data were analysed by a two-way repeated measure mixed model (ANOVA allowing missing values) including Geisser-Greenhouse correction to account for possible unequal variances between the test groups. Subsequently, post hoc comparison between test groups on every test day was performed with Dunnett’s correction for multiple comparisons when multiple test groups were tested against the positive control group, Sidak’s correction when only two groups were compared and Tukey for comparison between all test groups. In figure legends group sizes and F statistics are provided. Post hoc differences between negative and positive control group are indicated by * and differences between positive control group and inhibitors/knockout by # and $ on the figures. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Rotarod data are expressed graphically by individual data points and analysed by Kruskal-Wallis one-way ANOVA with Dunn’s post hoc comparison and described by median and 95% confidence intervals. CGRP release data are presented as individual measures and mean. Two-way repeated ANOVA was performed with comparison between test and control group at each concentration and between basal level and positive control within groups. Post hoc tests as appropriate. Ex vivo vasoactivity data are shown as mean percent of precontraction ± SEM and analysed by two-way repeated measure mixed model and Sidak’s post hoc comparison between experimental groups. All data were analysed, and graphs created in GraphPad Prism 9. Corrected P-values below 0.05 were considered statistically significant.
Results
Prostaglandins and TRPA1 are involved in GTN-mediated hypersensitivity
In the GTN mouse model, we tested the effect of ibuprofen 100 and 200 mg/kg IP and found that both doses were effective day 1 and 3 of the 9-day test paradigm. After that, ibuprofen did not inhibit GTN-induced hypersensitivity (Figure 1(a)). Overall difference between test groups F(3,42) = 38.5, P < 0.0001 and post hoc comparison show that the GTN group was significantly different from the vehicle group on all test days (P < 0.0001 all days) and both doses of ibuprofen inhibited this response on days 1 and 3 (P = 0.0008 – 0.006 days 1 and 3 and P = 0.17 – 0.99 days 5, 7 and 9). Chemical inhibition of TRPA1 by HC030031, F(3,43) = 28.9, P < 0.0001 and genetic deletion of Trpa1 F(3,42) = 33.9, P < 0.0001 were both fully effective against GTN throughout the test protocol (Figure 1(b) and (c)).
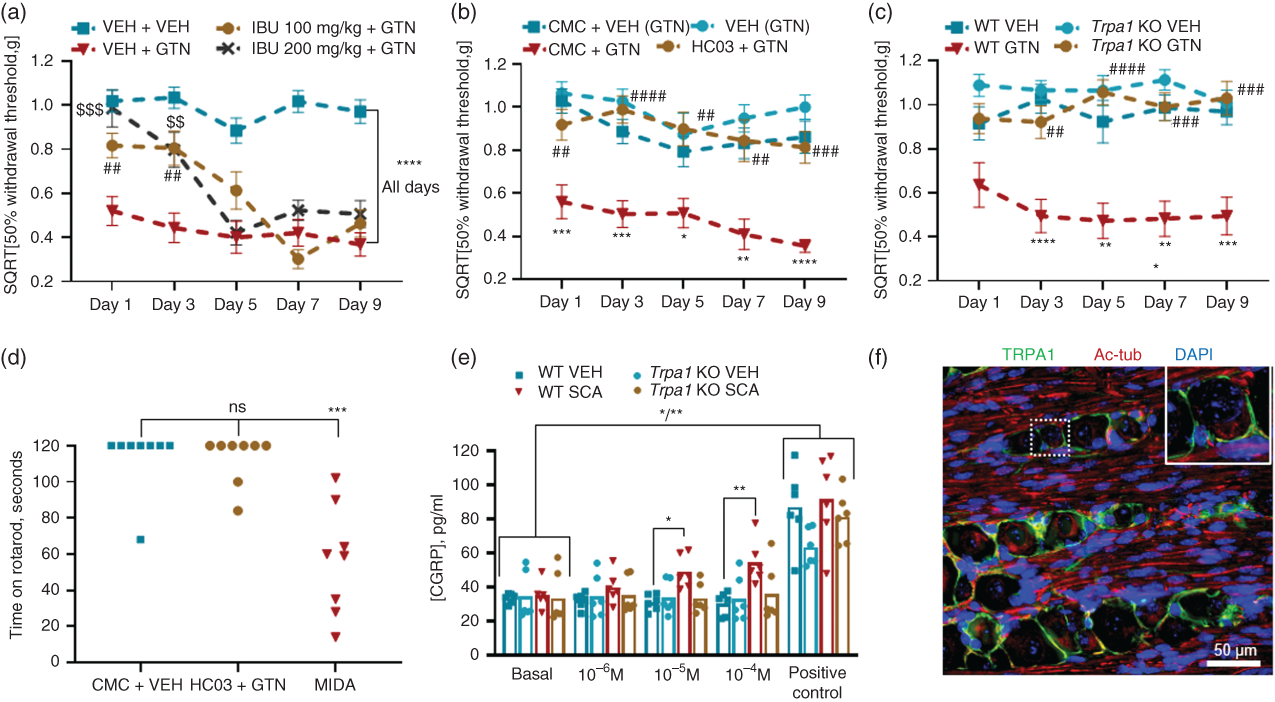
Prostaglandins and TRPA1 are involved in GTN-mediated hypersensitivity. (a–c) Sensitivity to tactile stimulation of the plantar area with von Frey filaments 2 h after GTN 10 mg/kg IP given on test days 1, 3, 5, 7 and 9 in combination with chemical inhibitors. Data shown as mean ± SEM of SQRT [50% withdrawal threshold in grams]. (a) Ibuprofen 100 and 200 mg/kg IP inhibited GTN-induced hypersensitivity on days 1 and 3. Overall treatment effect F(3,42) = 38.5, P < 0.0001, n = 11-12. (b) TRPA1 antagonist HC030031 100 mg/kg PO (dissolved in 0.5% carboxymethylcellulose, CMC in tap water) completely prevented the development of hypersensitivity throughout the protocol, F(3,43) = 28.9, P < 0.0001, n = 11-12 and (c) Trpa1 knockout mice were also protected from the effects of GTN, F(3,42) = 33.9, P < 0.0001, n = 11-12. (d) Rotarod performance 2 h after HC030031 + GTN treatment compared to vehicle controls and midazolam 1 mg/kg IP positive controls, Kruskal-Willis statistics 15.6, P = 0.0004, n = 8. (e) Individual data points and mean CGRP released from trigeminal ganglia of wild type and Trpa1 knockout mice stimulated with super cinnamaldehyde 10−6–10−4 M and capsaicin 10−5 as positive control, F(3,20) = 2.8, P < 0.06, n = 6. (f) Representative 3 µm paraffin section of trigeminal ganglion showing the presence of TRPA1 (green) in neurons. Co-stained for acetylated tubulin (red) and DAPI (blue).
Normal motor function following HC030031 + GTN treatment was confirmed as median time spent on the rotarod was 120 s in both the test (95% CI 84–120) and vehicle (95% CI 64–120) group P > 0.99 and 60 s (95% CI 14–102) in the midazolam positive control group P = 0.008 (Figure 1(d)).
Specific stimulation of trigeminal ganglia by TRPA1 agonist super cinnamaldehyde 10−6, 10−5 and 10−4 M resulted in 9% (P = 0.23), 51% (P = 0.011) and 69% (P = 0.0097) increased release of CGRP, respectively, compared to the vehicle control. In trigeminal ganglia from the Trpa1 null mice, super cinnamaldehyde did not stimulate CGRP release as exposure to 10−6, 10−5 and 10−4 M super cinnamaldehyde resulted in 11% (P > 0.99), −13% (P > 0.99) and 9% (P = 0.97) percent changes in CGRP release compared to vehicle (Figure 1(e)). Stimulation with positive control TRPV1 agonist capsaicin 10 µM resulted in 183–260% (P = 0.006–0.03) more CGRP in the four treatment groups, proving that all tissue samples had the ability to release CGRP.
Immunohistochemical staining confirmed TRPA1 localisation in neuronal cell membranes of the trigeminal ganglion (Figure 1(f)) as previously shown in dural nerve fibres (34) and trigeminal ganglion (13). These data suggest that CGRP and TRPA1 dependency of GTN-induced hypersensitivity is due to release of CGRP from trigeminal neurons following TRPA1 stimulation.
Cilostazol-induced hypersensitivity is independent of TRPA1 and prostaglandins but dependent on CGRP and KATP channels
In the next series of experiments, we first established a mouse model of cilostazol-induced migraine by administration of cilostazol 100, 300 and 500 mg/kg PO and evaluated the plantar sensitivity at 2, 4 and 6 h after gavage (Figure 2(a) and (b)). There was a significant overall treatment effect of cilostazol at 6 h, F(3,20) = 40.8, P < 0.0001 and both 300 and 500 mg/kg induced hypersensitivity. The effect was most prominent after 6 h (days 1 and 3 not shown) and this time point was tested onwards. The presence of cephalic hypersensitivity to stimulation with von Frey filaments was also demonstrated (Figure 2(c)), effect of treatment F(1,17) = 15.1, P = 0.001.

Cilostazol-induced hypersensitivity is independent of TRPA1 and prostaglandins but dependent on CGRP and KATP channels. (a–f) Sensitivity to tactile stimulation of the plantar or cephalic (c) area with von Frey filaments 6 h (2, 4 and 6 h in (b)) after cilostazol (CIL) 500 mg/kg PO dissolved in 0.5% CMC (100, 300 and 500 mg/kg in (a–b)). Data shown as mean ± SEM of SQRT [50% withdrawal threshold in grams]. (a) Cilostazol-induced hypersensitivity 6 h after gavage of 300 and 500 mg/kg, but not 100 mg/kg, F(3,20) = 40.8, P < 0.0001, n = 6. (b) The effect size was largest 6 h after treatment, F(3,20) = 6.7, P = 0.003, n = 6 (same mice as in (a)). Cilostazol also induced cephalic hypersensitivity (c), F(1,17) = 15.1, P = 0.001, n = 7–10. Trpa1 knockout mice were as sensitive as wild types to cilostazol (d), F(3,40) = 39.2, P < 0.0001, n = 10–12. Also, ibuprofen 100 mg/kg IP was ineffective against cilostazol-induced hypersensitivity whereas CGRP monoclonal antibody, ALD405 inhibited the response (e), F(3,43) = 18.2, P < 0.0001, n = 11–12. Last, in (f) we found that KATP channel blocker glibenclamide 1 mg/kg IP completely inhibited the effect of cilostazol while CGRP receptor antagonist olcegepant 1 mg/kg IP did so partially, F(3,44) = 22.1, P < 0.0001, n = 11–12.
The cilostazol model was characterised by use of the inhibitors known to be effective in the GTN model. Knockout of Trpa1 did not protect against the effect of cilostazol as these mice were as sensitive as wild type controls (Figure 2(d)), P = 0.09 day 1 and 0.93 – 1 days 3 to 9. Ibuprofen was also ineffective in blocking the effect of cilostazol (Figure 2(e)), P = 0.14–0.84 days 1 to 9. The monoclonal antibody against CGRP, ALD405, was given on day 0 and prevented hypersensitivity following cilostazol treatment, almost reaching the level of the negative control until day 7 where antibody titer had declined (27) and the effect size became smaller, P = 0.045–0.001 days 1 to 5 and P = 0.12–0.35 days 7 and 9. CGRP involvement in cilostazol-induced hypersensitivity was also indicated by partial efficacy of CGRP receptor antagonist olcegepant. The effect disappeared day 7 and 9 for unknown reasons (Figure 2(f)), P = 0.006–0.04 days 1 and 5 and P = 0.15–0.55 days 3, 7 and 9. The combination of olcegepant and cilostazol had no effect on motor function. Median time on the rotarod was 120 s for both the negative control group (95% CI 80–120) and the test group (95% CI 84–120), P > 0.99, whereas the median of the positive control was 38 s (95% CI 19–82), P = 0.0005 (Supplementary data, Figure 2). KATP channel blocker glibenclamide completely inhibited the effect of cilostazol throughout the protocol (Figure 2(f)), P = 0.002–0.046.
Levcromakalim-induced hypersensitivity is independent of TRPA1 and prostaglandins but dependent on CGRP
The characterization panel of inhibitors and genetic deletion used above was also applied to the mouse model of levcromakalim-induced migraine. All in all, levcromakalim provocation mirrored cilostazol provocation. First, levcromakalim was independent of TRPA1 (Figure 3(a)), as Trpa1 knockout mice did not have higher 50% withdrawal thresholds than the wild type positive control group, P = 0.47 – 1. Also, inhibition of prostaglandins by ibuprofen was ineffective with no elevation of sensitivity levels at any days (Figure 3(b)), P = 0.72 – 1. Target engagement of the KATP channel was confirmed by almost complete inhibition of hypersensitivity by glibenclamide (Figure 3(b)), as previously described (23). Olcegepant was partially effective, though only significant on day 9, P = 0.019 and days 1, 3, 5 and 7, P = 0.09–0.73 (Figure 3(b)) whereas ALD405 (Figure 3(c)) provided full protection from the effect of levcromakalim, P = 0.001–0.08. To verify the involvement of the CGRP receptor, we also tested Ramp1 null mice following injections with levcromakalim. These knockout mice had complete resistance to levcromakalim induced hypersensitivity on all test days (Figure (3d)), P = 0.02 – < 0.0001, proving that CGRP and its receptor are necessary mediators following the actions of levcromakalim. In support of the presented data measuring hind paw sensitivity, levcromakalim induced cephalic hypersensitivity to a degree similar to GTN (Figure 3(e)), overall treatment effect F(2,21) = 7.0, P = 0.005. Finally, we demonstrated that the combination of glibenclamide and levcromakalim did not differ from the negative control group on rotarod performance (Figure 3(f)). Medians were 120 s (95% CI 50–120) and 118 s (95% CI 65–120), respectively, P > 0.99.
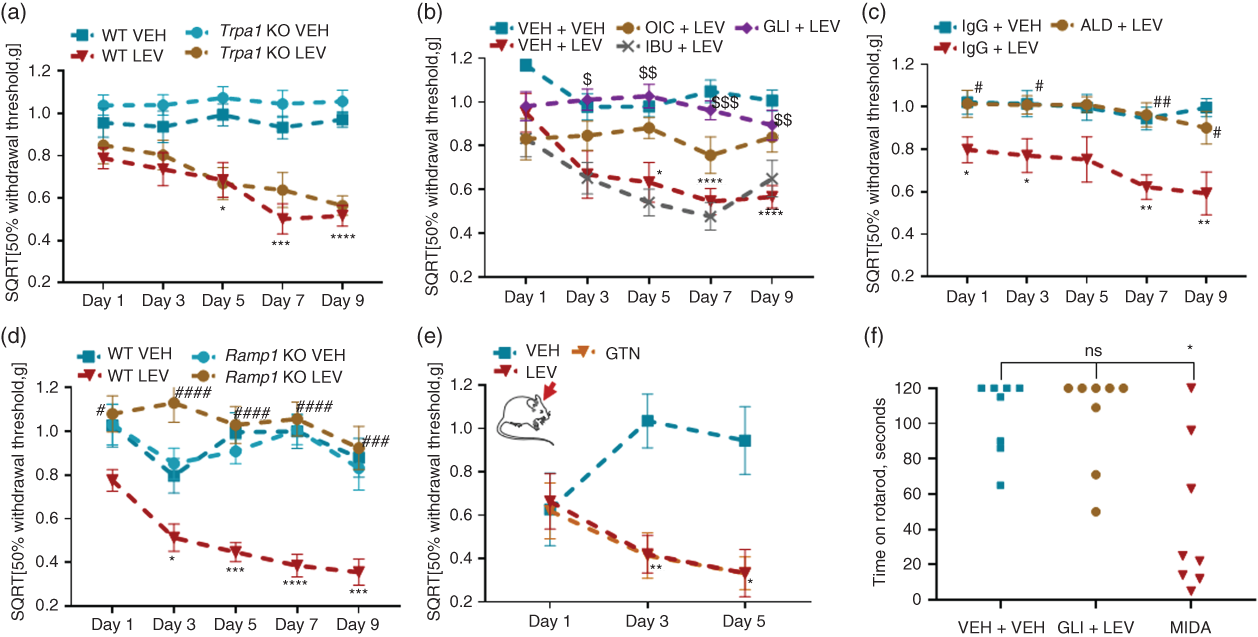
Levcromakalim-induced hypersensitivity is independent of TRPA1 and prostaglandins but dependent on CGRP. (a–e) Sensitivity to tactile stimulation of the plantar area (cephalic in (e)) with von Frey filaments 2 h after levcromakalim (LEV) 1 mg/kg IP given on test days 1, 3, 5, 7 and 9 in combination with chemical inhibitors. Data shown as mean ± SEM of SQRT [50% withdrawal threshold in grams]. (a) Trpa1 KO mice were sensitive to provocation of hypersensitivity by levcromakalim, F(3,50) = 28.5, P < 0.0001, n = 12–15 and ibuprofen 100 mg/kg IP did not inhibit the effect either (b). KATP channel blocker glibenclamide 1 mg/kg IP completely inhibited the effect of levcromakalim and CGRP receptor antagonist olcegepant 1 mg/kg IP did so partially (b), F(4,52) = 16.9, P < 0.0001, n = 9–12. CGRP monoclonal antibody, ALD405 fully antagonised levcromakalim (c), F(2,30) = 13.3, P < 0.0001, n = 9–12 and this result was supported by Ramp1 KO mice being resistant to the effect of levcromakalim (d), F(3,39) = 23.3, P < 0.0001, n = 10–12. In (e) cephalic hypersensitivity of levcromakalim is illustrated along GTN, F(2,1) = 7.0, P = 0.005, n = 7–8. (f) Rotarod performance 2 h after glibenclamide + levcromakalim treatment compared to vehicle controls and midazolam 1 mg/kg IP positive controls, Kruskal-Willis statistics 8.8, P = 0.013, n = 8.
Cilostazol- and levcromakalim-induced hypersensitivity is not dependent on direct release of CGRP from the trigeminal ganglion or the trigeminal nucleus caudalis
The findings that both cilostazol- and levcromakalim-induced hypersensitivity were blocked by CGRP neutralisation was surprising and not previously described. We investigated the phenomenon in two different ex vivo preparations. First, in a myograph bath, KATP channel-induced vasodilation by levcromakalim was downstream from CGRP because there was no inhibitory effect of 1 µM olcegepant on levcromakalim-induced vasodilation. Both the olcegepant- and vehicle-treated artery segments dilated to increasing concentrations of levcromakalim, with no difference between the two groups (Figure 4(a)), F(1,5) = 0.08, P = 0.79. Subsequent contraction by addition of glibenclamide was also the same in olcegepant and vehicle pre-treated segments, F(1,5) = 0.00004, P > 0.99. Next, we tested the ability of levcromakalim and cilostazol 10−7–10−4 M to directly stimulate release of CGRP from trigeminal ganglia and trigeminal nucleus caudalis. Cilostazol (Figure 4(b) and (c)) and levcromakalim (Figure 4(d) and (e)) did not stimulate CGRP release. The changes from vehicle were −14%, P = 0.91 for cilostazol and 26%, P = 0.52 for levcromakalim in TG. In TNC, the CGRP release compared to vehicle was −8%, P = 0.95 for cilostazol and 8% P = 0.97 for levcromakalim. In Supplementary data, Figure 4, different concentrations of cilostazol and levcromakalim are shown. Additionally, we tested the cAMP analogue 8-Br-cAMP and adenylate cyclase stimulator forskolin and these did not stimulate CGRP either (Supplementary data, Figure 4).

CGRP dependency of cilostazol- and levcromakalim-induced hypersensitivity is not caused by direct release of CGRP. (a) Dilation of basilar arteries shown as percent of precontraction to increasing concentration of levcromakalim in the presence of CGRP receptor antagonist olcegepant 1 µM or its vehicle. Olcegepant did not alter the response to levcromakalim, F(1,5) = 0.08, P = 0.79, n = 6. Subsequent addition of increasing concentrations of KATP channel blocker glibenclamide contracted the arteries, but was unaffected by olcegepant F(1,5) = 0.00004, P > 0.99, n = 6. (b–c) Release of CGRP from trigeminal ganglia (b), F(1,6) = 0.28, P = 0.61, n = 4 and brain stem (c) F(1,6) = 1.14, P = 0.33, n = 4 following stimulation with cilostazol 10−4 M and capsaicin 10−5 M as positive control. (d–e) As previous but stimulated with levcromakalim 10−4 M. Trigeminal ganglia (d), F(1,11) = 2.30, P = 0.16, n = 6–7 and brain stem (e) F(1,11) = 0.12, P = 0.74, n = 6–7. Note, the different range of the y-axis in (c) and (e) is due the use of rat and human CGRP ELISA kits, respectively.
Discussion
Our study provides new translational knowledge on signalling pathways involved following administration of the human experimental triggers of migraine GTN, cilostazol and levcromakalim. Unexpectedly, we found that both cilostazol- and levcromakalim-induced hypersensitivity was rescued by treatment with CGRP antagonising drugs despite CGRP acting upstream from these migraine triggers on single-cell level. This in vivo finding could not be replicated in ex vivo organ preparations of the trigeminal ganglion and the trigeminal nucleus caudalis, suggesting that a complex interplay between different tissues and cell types is involved in the CGRP signal following cilostazol and levcromakalim. Our findings and hypothesis of their location are illustrated in Figure 5.
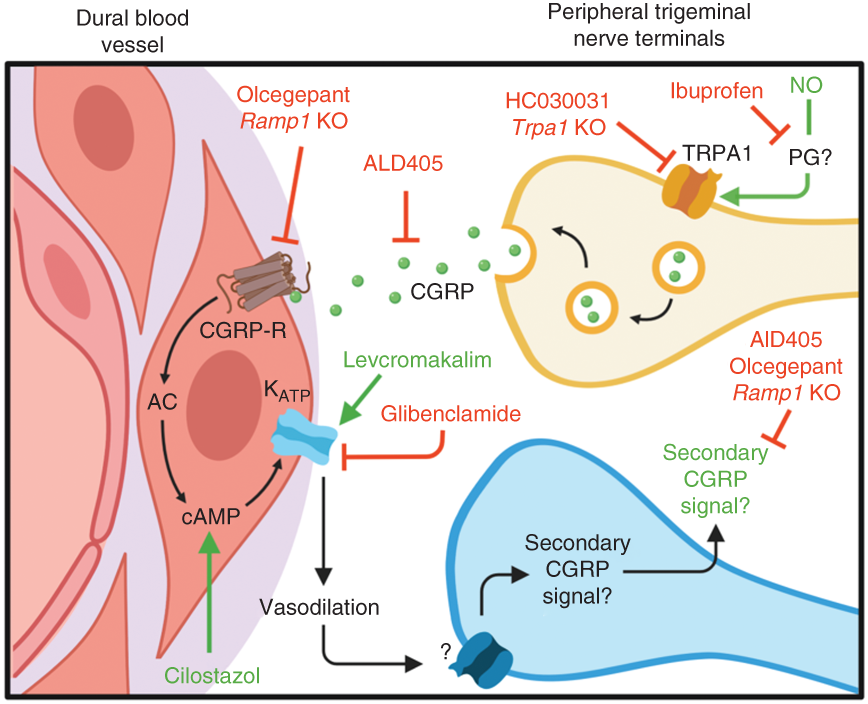
Schematic illustration of signalling pathways involved in GTN- cilostazol- and levcromakalim-induced hypersensitivity. Signalling pathways suggested to be involved in GTN-, cilostazol- and levcromakalim-induced hypersensitivity by data given in Figures 1–4. The CGRP signalling event downstream from cilostazol and levcromakalim is not previously described and cannot be explained by direct CGRP release upon stimulation of trigeminal ganglia or trigeminal nucleus caudalis by cilostazol and levcromakalim. Therefore, we suggest that crosstalk between meningeal arteries and trigeminal sensory fibres is necessary to generate the secondary CGRP signal.
TRPA1 involvement in GTN-, cilostazol- and levcromakalim-induced migraine-like pain
First, we show a clear difference between GTN and cilostazol/levcromakalim in terms of involvement of prostaglandins and TRPA1. The effect of GTN was completely inhibited by both chemical inhibition and genetic deletion of Trpa1. The same was found by Marone and colleagues (13), who further identified the interaction of TRPA1 and reactive oxygen species derived from GTN as responsible for the induced hypersensitivity. Trigeminal TRPA1 channels have also been identified as a target for nitroxyl (HNO), a redox product of NO (14). A similar phenomenon has been described in a model of muscle hypertrophy where the effect of NO was not attributed to its binding to sGC but to the generation of peroxynitrite subsequently targeting TRPV1 (15). The action of NO derivatives on TRPA1 fits well with the previously documented CGRP dependency of GTN animal models (27,35,36) and ex vivo CGRP release in both rat and mouse tissue following HNO stimulation of TRPA1 (37). Our data also showed that TRPA1 stimulation by the TRPA1 agonist super cinnamaldehyde caused release of CGRP from mouse trigeminal ganglia. In the rat model of GTN-induced migraine, TRPA1 involvement was also confirmed (38). However, others found that GTN-induced hypersensitivity was completely blocked by the sGC inhibitor ODQ (12). Thus, evidence points in two directions as to whether the TRPA1-CGRP-cAMP or the sGC-cGMP pathway mediates GTN-induced hypersensitivity. Perhaps both are involved, and the complete inhibitions observed in mouse models by different labs may be an expression of limited resolution of the model. Despite this controversy, the common denominator for both pathways is the KATP channel. We previously showed that KATP channel blocker glibenclamide provides complete inhibition of GTN-induced hypersensitivity (23).
Prostaglandin involvement in GTN-, cilostazol- and levcromakalim-induced migraine-like pain
GTN provocation also differed from cilostazol and levcromakalim provocations regarding the involvement of prostaglandins. Thus, ibuprofen inhibited GTN-induced hypersensitivity on the first two test days, whereas it was ineffective towards cilostazol and levcromakalim provocations. Why the effect of ibuprofen disappeared during the chronic protocol is unknown, but likely has to do with recruitment of prostaglandin-independent pathways mediating the basal hypersensitivity that is also present in the GTN mouse model (21,27) as documented in our supplementary material. Prostaglandin inhibition was also effective in the GTN – single injection – rat model (39) and prostaglandins can activate TRPA1 (40), suggesting that these are closely linked. TRPA1 and prostaglandin inhibition were both effective in the GTN model, but not in the cilostazol and levcromakalim models.
CGRP involvement in GTN-, cilostazol- and levcromakalim-induced migraine-like pain
Given that binding of CGRP to its receptor causes smooth muscle relaxation via cAMP and KATP channel opening (3,17), cilostazol and levcromakalim act downstream from CGRP in what we describe as the CGRP-cAMP- KATP axis. We have previously shown that GTN-induced hypersensitivity can be completely blocked by a CGRP antibody (27) but it was unexpected that also cilostazol- and levcromakalim-induced hypersensitivity could be blocked by inhibition of CGRP signalling; both by a small molecule CGRP receptor antagonist (olcegepant) and a monoclonal antibody against CGRP itself (ALD405). The effect of olcegepant was partial and variable indicating that a larger dose perhaps would have been better but efficacy of CGRP receptor antagonism was also shown by knockout of the rate limiting CGRP receptor component RAMP1 where efficacy was complete. This dependency of CGRP could not be explained by a direct release of CGRP from the trigeminal ganglion or the trigeminal nucleus caudalis following stimulation with cilostazol or levcromakalim. Our data is supported by a study in rats where levcromakalim did not stimulate CGRP release from neither dura mater, trigeminal ganglion nor trigeminal nucleus caudalis (31). Hence, CGRP released in vivo secondary to cilostazol or levcromakalim injection must require more complex crosstalk between different cell types than observed in the ex vivo organ preparations. We suggest that this crosstalk is between meningeal arteries and trigeminal sensory fibres as illustrated in Figure 5. Brain areas such as trigeminal nucleus caudalis, hypothalamus, thalamus or cortex are unlikely to be involved as the CGRP targeting antibodies hardly enter the central nervous system (CNS) (41) and, to the extent they do, are ineffective towards GTN-induced hypersensitivity (20). CGRP dependency of cilostazol-induced hypersensitivity could be explained by a limited substrate (cAMP) for cilostazol when there is no CGRP stimulation of the CGRP receptor. If there is very little cAMP, the PDE3 would not be active and thus inhibition of PDE3 by cilostazol would be irrelevant and mice would not become hypersensitive. This hypothesis makes the proposed secondary CGRP signal redundant, but it cannot explain the CGRP dependency of levcromakalim-induced hypersensitivity as levcromakalim acts directly on KATP channels independent of cAMP. Our data suggest that cilostazol- and levcromakalim- induced hypersensitivity employs similar signalling and therefore we do not support a hypothesis that only explains CGRP dependency of cilostazol. Another, possible mechanism of action for cilostazol and levcromakalim is hyperpolarisation of neurons by KATP channel opening and subsequent signalling for instance via hyperpolarization-activated cyclic nucleotide–gated (HCN) channels (42). However, this hypothesis is contradicted by the analgesic action of KATP channel openers in the CNS (43–46) and very limited expression of KATP channels in trigeminal ganglia of human and mouse (in house, submitted data).
Strengths and limitations
An important methodological point of discussion is the measurement of plantar versus periorbital/cephalic sensitivity to tactile stimulation. Despite a recent shift towards cephalic measurements being considered more relevant to migraine than peripheral measurements; this does not change the fact that GTN (12,21), cilostazol and levcromakalim induce both cephalic and plantar hypersensitivity in mice and that hypersensitivity respond to the migraine-specific drugs sumatriptan and olcegepant (12,21,27). The same is true for models inducing migraine-like pain via direct stimulation of the dura mater (47,48) and triptan-induced sensitisation (49). If only one is measured it does not mean the other is not present. The variance of testing the head is considerably larger than that of the paw. In complicated studies requiring lots of experimental animals, we therefore mainly rely on testing the paw and confirm that the face responds similarly. Experimental migraine studies often use several outcome parameters such as light sensitivity (50) or mouse grimacing (51). However, these methods have greater variance and do not always report equally over time (52). That poses a problem in a study such as the present where data need to be stable and comparable over time in order to dissect migraine signalling pathways. This view is reflected in the fact that the present study, despite testing mainly plantar sensitivity, used more than 600 mice. Spontaneous behavioural and other outcome parameters are very important in other types of studies, for instance studies of novel migraine provoking agents and studies of therapeutic efficacy of novel drugs.
Human and rodent model of provoked migraine
The mouse models applied here mirror the human model of provoked migraine because they use the same triggering compounds. Here, the triggers were applied repeatedly to ensure a proper effect window and to verify the results on individual days. The repeated injections resulted in a basal hypersensitivity (provided as supplementary data) that was present without simultaneous presence of trigger compounds. We have focused on the acute responses to antagonistic drugs and in all, but the ibuprofen treatment of GTN-induced hypersensitivity and the olcegepant + cilostazol combination, administration of antagonists along with the trigger was equally effective in the acute and chronic phase of the protocol. From this we learn that many of the mechanisms are shared between the acute and basal hypersensitivity.
However, responses to antagonistic drugs are not always the same in human and rodent models. Olcegepant and other CGRP receptor antagonists are effective in both the rat and mouse model of GTN-induced migraine (27,36,53), but olcegepant failed in the human model (54). One difference between rodent and human studies is the timing of olcegepant in relation to GTN. In rodents olcegepant was given prior to or simultaneously with GTN whereas in the human study it was given after, but before the onset of delayed headache. Another important difference is the route of administration and dose of GTN. A much higher dose of GTN is given to rodents to induce migraine-like changes and it is unknown how 10 mg/kg IP as a bolus correlates to the human IV 20 min infusion at 10 µg/kg. In rodents, involvement of TRPA1 and CGRP is essential, but maybe the 1000-times lesser dose in humans does not activate the TRPA1-CGRP pathway – but only sGC, thereby bypassing CGRP signalling. However, Ramachandran and colleagues also found effect of olcegepant in rats when using IV infusion of GTN 80 µg/kg, but found that pre-treatment was better than post-treatment (35). Thus, the reason for the discrepancy between the human and rodent models of provoked migraine is not clear. Importantly, in this case and for sumatriptan (55), the rodent models seem to predict responses in spontaneous migraine better than the human GTN model.
The present study shows that blocking cAMP degradation and opening KATP channels to induce migraine-like pain are not CGRP independent in mouse models. Why CGRP is involved secondary to cAMP and KATP activation is so far unexplained. The dilation of meningeal arteries following GTN (56) (via the TRPA1-CGRP axis), cilostazol (57) and levcromakalim (58) may be sensed by trigeminal sensory nerves and this signal may result in subsequent release of CGRP. The data suggest a feed-forward loop in which CGRP could induce its own release via its vasodilatory effect (59).
Key findings
CGRP signalling is identified as essential in mouse models of GTN-, cilostazol- and levcromakalim-induced migraine. CGRP dependency of cilostazol- and levcromakalim-induced hypersensitivity cannot be replicated ex vivo, suggesting the phenomenon to be caused by complex crosstalk between different tissues/cell types. TRPA1 channels are involved in GTN-induced hypersensitivity, whereas cilostazol- and levcromakalim-induced hypersensitivity bypasses TRPA1.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024211038884 - Supplemental material for CGRP-dependent signalling pathways involved in mouse models of GTN- cilostazol- and levcromakalim-induced migraine
Supplemental material, sj-pdf-1-cep-10.1177_03331024211038884 for CGRP-dependent signalling pathways involved in mouse models of GTN- cilostazol- and levcromakalim-induced migraine by Sarah L Christensen, Rikke H Rasmussen, Charlotte Ernstsen, Sanne La Cour, Arthur David, Jade Chaker, Kristian A Haanes, Søren T Christensen, Jes Olesen and David M Kristensen in Cephalalgia
Footnotes
Author contributions
SLC and DMK designed the experiments with advice from JO. SLC and CE performed in vivo experiments and data analyses. RHR, SLaC, KAH, AD and JC did ex vivo experiments and data analysis. STC made the immunofluorescence. SLC and DMK interpreted the data. SLC, DMK and JO wrote the manuscript with contributions from all other authors.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Candys Foundation and Læge Sofus Carl Emil Friis og Hustru Olga Doris Friis' Legat.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.