
Abstract
Background
Headache and Neurological Deficits with cerebrospinal fluid (CSF) Lymphocytosis (HaNDL) is an increasingly recognised syndrome but the aetiology remains unclear. HaNDL has striking clinical features similar to Familial Hemiplegic Migraine (FHM), commonly related to gene mutations encoding the P/Q-type voltage-gated calcium channel (VGCC).
Case report
We report a case of HaNDL associated with high P/Q-type voltage-gated calcium channel antibodies. Extensive investigations excluded alternative diagnoses and CSF lymphocytosis resolved within 3 months. The case was complicated by raised intracranial pressure resulting in an enlarged blind spot, papilloedema and bilateral lateral rectus palsies.
Conclusion
This novel association of P/Q-type voltage-gated calcium channel antibodies with HaNDL has implications for the pathology of HaNDL and spectrum of voltage-gated calcium channel-antibody disorders. We compare the clinical features of FHM and HaNDL and the potential pathological role of these antibodies. This case also highlights that raised intracranial pressure is a common feature of HaNDL, rarely resulting in serious complications.
Introduction
Headache and neurological deficits with CSF lymphocytosis (HaNDL), also termed pseudomigraine with CSF pleocytosis, is a self-limiting syndrome of migraine-like attacks (1,2). HaNDL has a striking similarity to Familial Hemiplegic Migraine (FHM); individuals experience recurrent episodes of headache with unilateral sensorimotor deficits with or without vision and speech disturbances lasting hours. Unlike FHM, however, episodes usually remit within weeks to months and do not recur. Other clinical differences are summarised in Table 1.
Clinical characteristics of FHM-1, HaNDL and migraine with aura (MA).
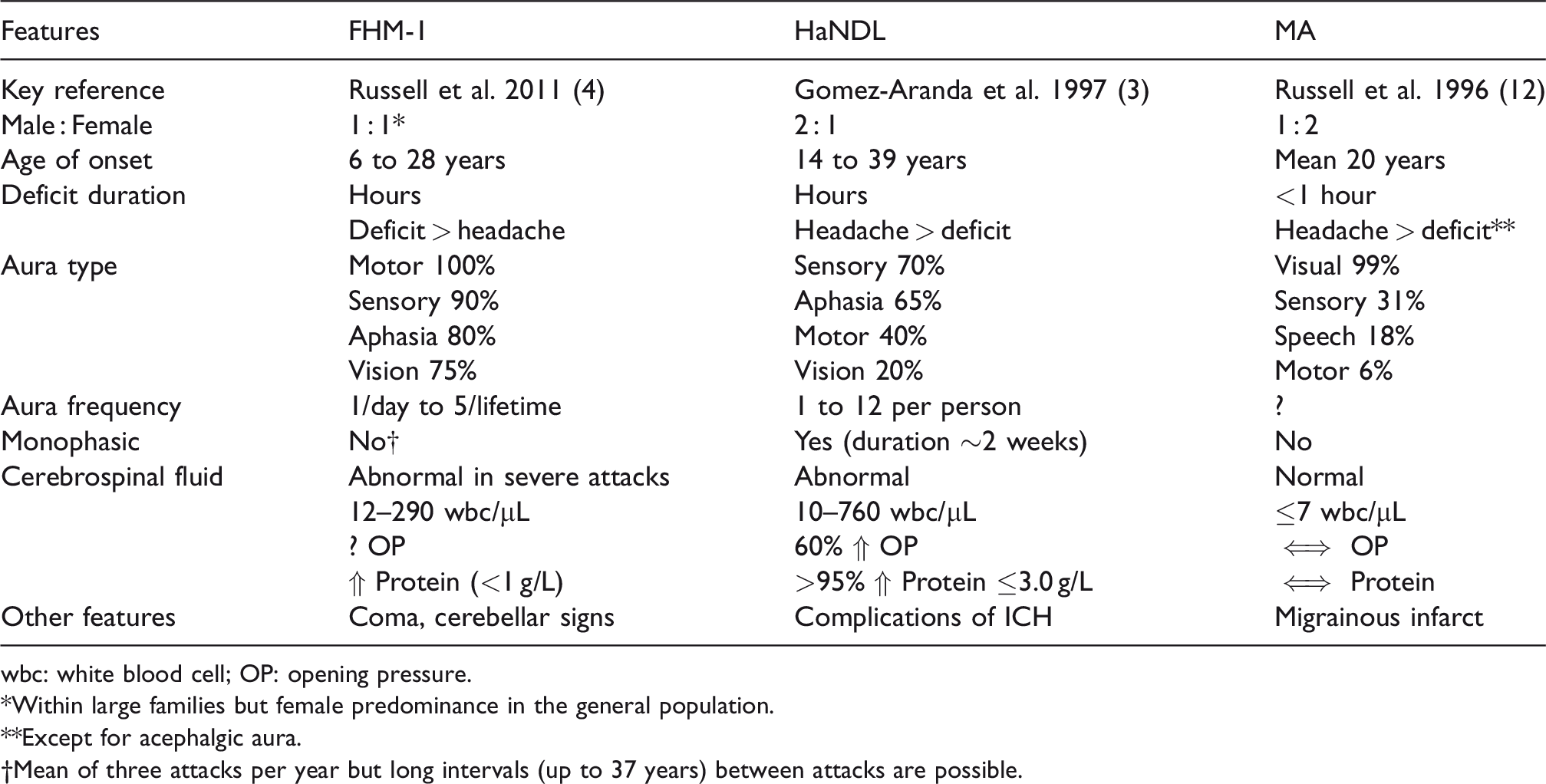
wbc: white blood cell; OP: opening pressure.
*Within large families but female predominance in the general population.
**Except for acephalgic aura.
†Mean of three attacks per year but long intervals (up to 37 years) between attacks are possible.
The first case series of HaNDL described increased CSF protein and intracranial hypertension (ICH) in addition to lymphocytosis (1). Transient non-epileptiform electroencephalography (EEG) changes are recognised as a feature of HaNDL (3). Neuroimaging is generally normal, except for transient focal cortical oedema and hypoperfusion usually corresponding to the neurological deficit (3). Cerebral angiography has been reported to precipitate both HaNDL and FHM attacks (3,4). FHM can also be triggered by head injury. Forty percent of FHM experience encephalopathic attacks, rarely complicated by fatal cerebral oedema (4). Encephalopathy as the core phenotype of HaNDL is unusual; however, there are reports of ‘malignant’ HaNDL with complications of ICH (5).
FHM is associated with mutations in ion-channel genes: CACNA1A, ATP1A2 and SCN1A. FHM1 is caused by mutations in CACNA1A encoding the α1-subunit of P/Q type voltage-gated calcium channels (VGCC) (Figure 1). There are numerous VGCC subtypes due to multiple genes encoding α1-subunits with different properties allowing specialised roles in neuronal subtypes.
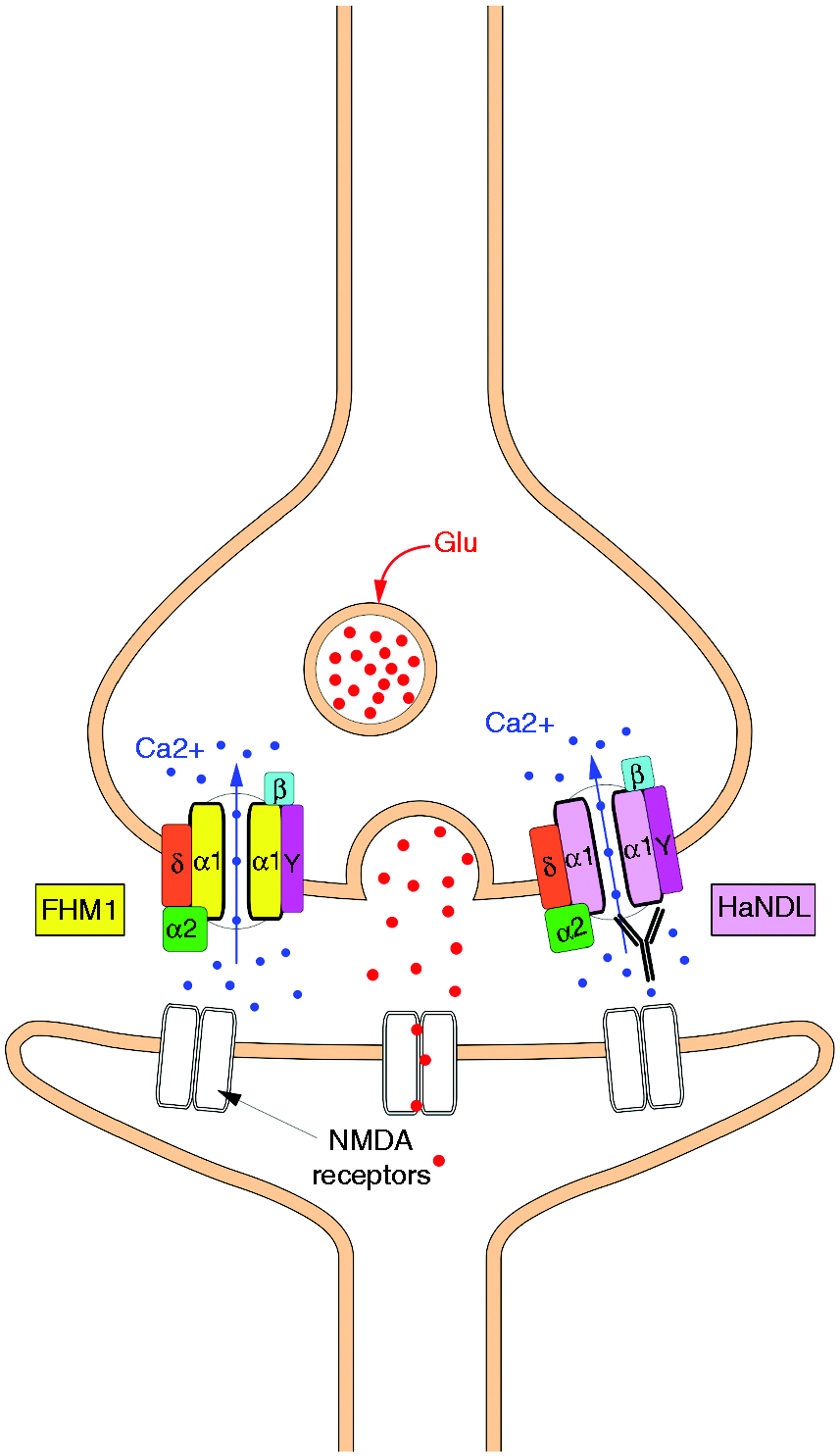
VGCC complex has α1-subunit ion conducting pore and α2δ, ɣ and β regulatory subunits. P/Q channel at presynaptic membrane is activated by high voltage depolarization. Calcium influx mediates vesicle release of neurotransmitters such as glutamate (Glu). Activated post-synaptic ligand-gated ion channels, such as NMDA receptors, lead to membrane depolarization and action potential propagation. (a) In FHM1, mutations within α1-subunit result in gain of function, probably due to negative shift of voltage-dependent activation leading to reduced threshold for action potential propagation. (b) In HaNDL, putative pathogenic VGCC-antibodies could result in gain of function by disrupting regulatory properties of the VGCC subunits.
Although the aetiology of HaNDL remains unknown, an ion channelopathy has been suggested. Chapman et al. (2003) searched for known FHM mutations and exonic polymorphisms within CACNA1A in eight HaNDL cases without success (6). Another study of four HaNDL cases screened for ion channel autoantibodies, demonstrated two cases with high antibody titers to T-type VGCC (CACNA1H) but not P/Q-type or other auto-antibodies (7).
Here we identify high P/Q-type VGCC antibodies in a typical patient with HaNDL. The presentation was characterised by confusion and ICH resulting in bilateral sixth nerve palsies. We discuss the potential pathogenic role of VGCC antibodies and the implications for the spectrum of VGCC-antibody related neurological diseases.
Case history
In February 2015, a 21-year-old right-handed female was admitted to hospital following multiple presentations to the emergency department (ED) with fluctuating headache and transient neurological symptoms. Her past medical history was notable only for migraine with visual aura since early teens occurring twice per year. There was no notable family history.
She first presented with 3 weeks of throbbing headache with photophobia and nausea, and an hour-long episode of migratory onset left-sided paraesthesia with difficulty in standing. A CT head was unremarkable. She received analgesia and was discharged symptom-free. Two days later she re-presented with headache recurrence and right leg weakness, resolving over 6 hours in ED. In Neurology clinic the following day, she reported a mild headache only and examination including fundoscopy was normal. She was initiated on amitriptyline but returned to the ED 10 days later with worsening headaches, new onset confusion and further right arm and face hemisensory disturbance, lasting 90 minutes.
On examination, her vital signs were normal. She was orientated but bradyphrenic (MMSE score 28/30). Fundal examination revealed bilateral papilloedema. Cranial nerve exam demonstrated bilateral lateral rectus palsies. Neck stiffness was absent and the remainder of the neurological examination was normal. Formal ophthalmic review confirmed bilateral lateral rectus weakness resulting in an esotropia with 12° and 25° prism dioptre for near and far vision, respectively. Visual acuities and Goldman fields were normal with the exception of an enlarged left blind spot.
Investigations
Routine blood tests, chest radiograph, MRI brain with contrast and venogram were all normal. Urinaryβ-HCG was negative. EEG revealed asynchronous frontotemporal bursts of delta waves, more prominent on the left with a slow background.
CSF examination revealed an opening pressure over 40 cm H2O, 310 white blood cells/μL (95% lymphocytes), protein 0.52 g/L, and glucose 3.5 g/L (serum 5.4 g/L). Oligoclonal bands were negative. CSF bacterial cultures, cryptococcal antigen test, mycoplasma pneumonia/listeria PCR tests were all negative. CSF viral PCR for enterovirus, mumps, HSV-1 and 2, VZV, parechovirus, EBV, CMV, JC, BK, measles, HHV-6, and HHV-7 were negative. CSF cytospin revealed morphologically normal lymphocytes.
Serology was negative for borrelia, toxoplasmosis, HIV, coxiella burnetti and bartonella. P/Q-type VGCC antibodies were 607 pmol/L (normal < 45). NMDA receptor (NMDAR), voltage-gated potassium complex channel (VGKC) antibodies, ANCA, ANA and rheumatoid factor were negative.
Management and clinical course
During her admission, she had a further episode of right hemisensory disturbance lasting 5 hours. She completed 2 weeks of intravenous acyclovir and was symptom free on discharge. CSF at 10 days, 3 weeks and 3 months after the first CSF revealed opening pressures of 40, 21 and 20 cm H2O, white cells/μL 208, 100 and 7, and protein 0.42, 0.40 and 0.16 g/L, respectively. She initially required prism correction. Ophthalmology follow-up revealed improvement of papilloedema and ophthalmoplegia at 1 month with complete resolution at 2 months. One year after presentation, she remained headache-free and VGCC antibodies were negative. Genetic testing for FHM was not performed.
Discussion
This is the first description of HaNDL in association with P/Q-type VGCC antibodies. This case also highlights that ICH is a common feature of HaNDL (3) and can, rarely, result in papilloedema, visual field defects and ophthalmoplegia (5). The pathogenesis and incidence of HaNDL is unknown and it remains a diagnosis of exclusion. Establishing an association between HaNDL and autoantibodies may improve diagnosis and avoid unnecessary treatment and investigations. Additionally, it would provide pathological insights into HaNDL and FHM.
As demonstrated here, HaNDL can be mistaken for migraine with aura. About a quarter of patients have a history of migraine (3). Aura in HaNDL is typically recurring and complex, consisting of hemisensory disturbance with motor or speech deficits lasting hours (3). HaNDL can be indistinguishable from FHM attacks except that motor weakness is not invariable and the headache outlasts the neurological deficit. Less common presentations of HaNDL include confusion, brainstem deficits and seizures (3). Examination between attacks is reportedly normal; however, this case demonstrates that signs of ICH can be detected.
When sought, CSF oligoclonal bands are universally absent in HaNDL (3). Associations with neuronal antibodies directed at Purkinje cell nuclei (7) and DNA repair proteins (8) have been reported. A single HaNDL case with NMDAR antibodies was reported but described atypical features with encephalopathic presentation and persistent neuropsychological deficits (9). Furthermore, the authors did not find NMDAR antibodies in archived CSF and sera of 12 HaNDL cases (9).
Only two studies consisting of a total of nine HaNDL patients have systematically searched for ion channel antibodies. The first identified two of four HaNDL cases with high T-type (CACNA1H) titers using enzyme-linked immunosorbent assay. Radioimmunoassay for P/Q-type VGCC antibodies, as used here, was negative in the HaNDL cases (7). The second repeated identification of T-type antibodies in addition to L-type (CACNAB1) antibodies to pooled patient sera using protein macroarrays (8). More sensitive cell-based assays for different VGCC subtypes, as used in detection of VGKC autoimmune encephalitis (10), have not yet been established. The various VGCC antibody associations may reflect heterogeneous pathological mechanisms underlying HaNDL.
This is the first case to be associated with unequivocally high titers of antibodies to P/Q-type VGCC containing α1-subunit encoded by CACNA1A. These antibodies are classically associated with Lambert-Eaton myasthenic syndrome (LEMS) (>85% of cases) and paraneoplastic cerebellar degeneration (40%), often in the context of small cell lung cancer (SCLC) (11). VGCC antibodies may be detected in 3–5% of patients with SCLC without LEMS but not in other populations (11). Our case of HaNDL did not have features of VGCC-related disorders, although electromyography was not performed. Repeat VGCC antibodies were negative, raising the possibility of a transient immune response rather than SCLC. A quarter of HaNDL cases report a viral prodrome (3). A prodrome was absent here and extensive viral tests were negative.
In FHM-1, CACNA1A mutations induce gain-of-function of P/Q VGCC with enhanced calcium influx and reduced threshold for cortical spreading depression (4). Given the clinical similarities between HaNDL and FHM-1, we propose that the VGCC antibodies in this case of HaNDL are likely pathogenic, leading to increased P/Q VGCC function. In LEMS, however, P/Q VGCC antibodies reduce pre-synaptic ion channel activity by cross-binding resulting in reduced calcium influx and neuromuscular junction acetylcholine release (11). As is the case with the VGKC-complex radioimmunoassay (10), it is possible that the pathogenic antibodies are associated with other proteins in complex with P/Q-type VGCCs, and enhance channel function indirectly rather than binding the channel itself.
The associations of VGCC antibody subtypes with HaNDL need replicating and their mechanisms investigated in vitro and in vivo. If replicated, VGCC antibodies will be of diagnostic value and may prompt trials of immunotherapy in severe HaNDL cases. This report suggests a potentially novel association of P/Q-type VGCC autoantibody with HaNDL, perhaps with an antigenic target that differs to LEMS. This may indicate a wider spectrum of VGCC than previously thought, with pathological implications.
Clinical implications
There is a novel association of HaNDL with unequivocally high P/Q-type VGCC antibodies. HaNDL has striking clinical similarities to FHM caused by mutations in gene encoding P/Q-type VGCC. Raised intracranial pressure is a common feature of HaNDL, rarely resulting in serious complications.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.