
Abstract
Introduction
Clinically, calcitonin gene-related peptide antagonising drugs are recognized as effective in migraine treatment, but their site of action is debated. Only a small fraction of these compounds pass the blood-brain barrier and accesses the central nervous system. Regardless, it has been argued that the central nervous system is the site of action. Here, we test this hypothesis by bypassing the blood-brain barrier through intracerebroventricular injection of calcitonin gene-related peptide antagonising drugs.
Methods
We used the glyceryl trinitrate (GTN) mouse model, which is well validated by its response to specific migraine drugs. The calcitonin gene-related peptide receptor antagonist olcegepant and the calcitonin gene-related peptide monoclonal antibody ALD405 were administered either intraperitoneally or intracerebroventricularly. The outcome measure was cutaneous mechanical allodynia.
Results
Mice given olcegepant intraperitoneally + GTN on day 1 had a mean 50% withdrawal threshold of 1.2 g in contrast to mice receiving placebo + GTN, which had a threshold of 0.3 g (p < 0.001). Similarly, in the ALD405 + GTN group, mice had thresholds of 1.2 g versus 0.2 g in the placebo + GTN group (p < 0.001). However, both drugs were ineffective when delivered intracerebroventricularly, as control and active groups had identical mechanical sensitivity thresholds, 0.2 g versus 0.1 g and 0.1 g versus 0.1 g for olcegepant and ALD405, respectively (p > 0.99 in both cases).
Discussion
The site of action of olcegepant and of the monoclonal antibody ALD405 is outside the blood-brain barrier in this mouse model of migraine. It is likely that these results can be generalised to all gepants and all antibodies and that the results are relevant for human migraine.
Introduction
Is migraine a cerebral or extracerebral disorder? The debate has pulsated for decades (1–5). Going back a few years in time, central mechanisms, for example brainstem activation were dominating for the site of origin for migraine without aura (6–8), but more recently the pendulum has been swinging back towards extracerebral mechanisms (9–11). The verdict is still out and therefore it is important to understand where different migraine mechanisms are located.
In this regard, localization of the effect of CGRP and of CGRP antagonism in migraine is of special interest. CGRP can induce migraine attacks in migraine sufferers (12) but it does not cross the blood-brain barrier in significant amounts and does not dilate brain arteries. Contrarily, the middle meningeal artery, which is not protected by the blood-brain barrier, is dilated by CGRP (13,14). Only a fraction of small molecule CGRP receptor antagonists penetrate the blood-brain barrier (15), and it has been suggested that the relatively high doses needed for efficacy reflect the need for a certain amount to penetrate into the CNS (1). A similar argument pertains to CGRP/CGRP receptor antibodies, which are much bigger and therefore even less able to cross the blood-brain barrier (4,5).These drugs are used for prophylaxis, which means that they circulate for weeks, leaving ample time for even the minute fractions that cross the blood-brain barrier to accumulate in the intracerebral tissues. Therefore, the antibodies could also, in theory, exert their action against migraine in the brain (16).
It is difficult to study the site of action of CGRP-signalling antagonists in humans. Here, we used a validated mouse model of migraine (17–19) to examine the effect of a small molecule CGRP receptor antagonist, olcegepant, and a CGRP neutralizing antibody, ALD405, given either systemically or intracerebroventricularly (ICV) in a blinded, vehicle-controlled design (double-blind). Our data strongly suggest that the site of action of both molecules is in the periphery.
Methods
Animals
Mouse experiments were performed under license 2017-15-0201-01358 from the Danish Animal Experiments Inspectorate and carried out in accordance with the ARRIVE guidelines. Mice were housed in a temperature (24°C) and humidity (45–55%) controlled room with a 12-hour light cycle, light on at 07.00 h, and free access to food (Altromin 1314, GmbH & Co., Germany and sunflower/corn/peanut mix, Brogaarden, Denmark) and tap water. All mice were adult, male C57Bl/6J in the age range 8–15 weeks. All handling and experimental procedures were performed between 08.00 and 16.00 h.
For systemic administration of olcegepant and ALD405, 26 naïve mice were used. These were delivered from Taconic, Denmark and group housed in Euro standard type 3 cages (Techniplast, Italy). Cages were enriched with two separate opaque red polycarbonate shelters – one tunnel and one igloo (Molytex, Denmark), a hemp rope (Fyns kran udstyr A/S, Denmark) suspended from the lid, nesting material (Happi-Mats and Enviro-Dri, Brogaarden, Denmark) and biting stick (Aspen Chew Bricks, Labodia, Switzerland). Cages were floored with sawdust (Aspen chip, Tapvei, Estonia).
Mice for ICV injections were delivered from Charles River, France, surgically prepared with a cannula (C315GS-4/Spc, 2.3 mm in length, Plastics One, Bilaney, Gernmany) in the right lateral ventricle (stereotactic coordinates from bregma; anterior-posterior −0.22 mm, lateral 1 mm, dorso-ventral −2.3 mm). Twenty-one mice were used 35 days after surgery and following 15 days of recovery from previous ICV injections of levcromakalim (an experiment looking at antinociceptive effect of central KATP channel opening). To avoid injury to the ICV cannulas, mice were single housed in plastic cages, L28 × W19 × H14 cm (SAMLA, IKEA, Denmark) with corresponding clear plastic lids prepared with 40 holes (2 mm in diameter) for ventilation. Boxes were floored with sawdust and enriched with two types of nesting material, biting sticks and an opaque red polycarbonate tunnel as described above. Water was provided by hydrogel water (solid drink, Triple A trading, Netherlands) in a small glass bowl (GLASIG, IKEA, Denmark) as the cages could not be fitted with a normal water bottle. Food was the same as for the naïve mice but supplemented with a daily portion (approximately 0.5 g) of nut paste (Nutella, Ferrero, Italy) to support animal weight. ICV mice were weighed every day and injected with saline (10 ml/kg) during anaesthesia and in case of weight loss.
GTN mouse model
The glyceryl trinitrate (GTN) mouse model is well validated by its response to migraine-specific drugs (17,19). We applied the model as described previously (19), but the number of GTN exposures was reduced to two per experiment. In short, GTN 10 mg/kg (Cambrex Germany, via the Capital Region Hospital Pharmacy, Denmark) was administered intraperitoneally (IP) and mechanical sensitivity was measured 2 hours later with von Frey filaments (Ugo Basile, Italy) on the plantar surface of the left hind paw using the up-down method (20) and an improved freely available online algorithm for calculation of 50% withdrawal thresholds (21). The experimenter was blinded to treatment group. GTN (5 mg/ml in ethanol) was diluted in saline to 1 mg/ml and a final alcohol concentration of 12.8%. The same vehicle was given to control mice in a 10 ml/kg volume. Prior to GTN, mice had received olcegepant HCl (MedChemTronica AB, Sweden) either IP or ICV and ALD405 (kindly donated by Alder Pharmaceuticals) IP or ICV in a subsequent experiment.
Intracerebroventricular injections
For ICV injections, mice were briefly placed under isoflurane anaesthesia (4% induction, 1.5% maintenance and a flow of 0.8-1 L/min of atmospheric air) and eyes were protected with ointment (viscotears 2 mg/g, Bausch + Lomb, Germany). Working aseptically, drugs were infused in a volume of 2 μl over 5 minutes using a 10 μl Hamilton syringe and the injection cannula was kept in place for another 2 minutes to avoid backflow before the dummy cannula was placed back and the animal returned to its home cage. Post-mortem, heads with the ICV cannula in place were fixed in 4% PFA and subsequently injected with Evans blue to confirm localisation of the cannula in the lateral ventricle.
Dose selection for olcegepant and ALD405
For the systemic administration of olcegepant and ALD405, IP doses of 1 mg/kg and 10 mg/kg, respectively, were selected based on previous experiments showing the effect of these doses in the GTN mouse model (19). The ICV dose of olcegepant, 0.45 µg/mouse, was based on the available literature on CNS availability of the gepants and reported efficacy against CGRP-induced light sensitivity of olcegepant 0.5 nmol/mouse ICV (22), equal to 0.45 µg/mouse olcegepant HCl. ALD405 was dosed at 10 µg/mouse, which equals 3.3% of the known effective dose given IP (300 µg/mouse). This is a factor 10 times larger amount than is found in the brain and CSF following systemic administration of antibodies (4,5). The vehicle for olcegepant was saline and PBS for IP and ICV injections, respectively.
Study protocol
Figure 1 provides an overview and timeline of the complete study protocol.

Study design.
Olcegepant experiment
Mice were allowed 14 days of washout from former injections. Prior to exposure, hind paw withdrawal thresholds were baseline tested. Twenty-one ICV mice were divided into two groups: Olcegepant + GTN (n = 11) and vehicle + GTN (n = 10), based on their baseline threshold and previous treatment. Mice that had previously received levcromakalim were equally distributed in the two groups. The 26 naïve mice for systemic administration of olcegepant were randomized into three groups: Vehicle for olcegepant + vehicle for GTN (n = 6), olcegepant + GTN (n = 10) and vehicle for olcegepant + GTN (n = 10) with respect to balancing their baseline values. Next day, referred to as day 1, mice received ICV and IP injections. Olcegepant or vehicle was injected 5–10 minutes before GTN. Mechanical sensitivity was measured 2 hours after injection of GTN. The day 1 paradigm was repeated on day 3.
ALD405 experiment
Before subsequent experiments with ALD405, mice were allowed to rest on days 4–7. On day 7, a baseline measure was taken, and ICV mice were randomized into two new groups: ALD405 + GTN and IgG control + GTN, balanced according to their baseline and whether they received olcegepant or vehicle in the first experiment. The same applied to the IP mice that were divided into three test groups: ALD405 + vehicle for GTN (n = 6), ALD405 + GTN (n = 10) and IgG control + GTN (n = 10). The CGRP antibody and IgG control antibody were administered ICV or IP on day 8. The next day animals were baseline tested, dosed with GTN and tested again 2 hours later. The GTN challenge was repeated day 11. Last, on day 13 the ICV mice were divided into two groups: olcegepant IP + GTN and vehicle for olcegepant IP + GTN to provide a positive control for the systemic effect of olcegepant in the ICV group of mice.
Drop outs
Throughout the study, 5/21 ICV mice were fully or partially excluded from the study, see Table 1 for all details. One mouse suffered an injury in relation to the ICV injections of vehicle on day 1, causing loss of body tonus and reflexes of the right hindleg. Another mouse, also from the vehicle group, presented with white cerebrospinal fluid on day 3. These mice were euthanized immediately, and all data withdrawn from the study. Another mouse was excluded on day 9 due to acute illness, but data prior to this point were included. Two mice were completely excluded due to concerns regarding placement of the guide cannula as the post-mortem injection of Evans blue in the ventricles was unsatisfactory.
List of mice used to complete the study. Individual mice are represented by their ID, ICV indicating a mouse implanted with an ICV guide cannula. For naïve mice, letters indicate separate home cages. Within the treatment columns the term negative control is used to differentiate from the test groups, but negative control mice received olcegepant vehicle or ALD405 vehicle + GTN. In the last column, day and reason for exclusion is given for individual mice.

Statistics
Data are presented as mean ± SEM of individual square root transformed (SQRT) 50% withdrawal thresholds for improved normal distribution, allowing parametric testing. Statistical analyses were performed in GraphPad Prism 8 (Graph Pad Software Inc., CA, USA) by two-way repeated measure ANOVA including Geisser-Greenhouse correction to account for possible unequal variances between the test groups. Analyses were followed by post-hoc comparison between all test groups adjusted with Bonferroni’s correction for multiple testing. Only relevant group comparisons within ICV groups and within IP groups are shown. Student’s t-test was used to compare the two groups in the positive control experiment. F-statistics and two-tailed p-values are reported and p < 0.05 was considered statistically significant. Group sizes were based on previous work with the same model (19), where group sizes of 10–12 provided adequate power to detect the effect of olcegepant and ALD405 at high levels of significance. Here, we aimed for 10 in each test group and six in the negative control group; that was used only to demonstrate that the assay is working.
Results
The site of action of olcegepant is outside the blood-brain barrier
We initially compared efficacy of olcegepant administrated by ICV and IP injections on GTN-induced allodynia (Figure 2). Overall, the ANOVA test revealed significant differences between treatment groups (F(4, 38) = 42.07, p < 0.001). At baseline, prior to administrations, the cannulated ICV groups had slightly lower (0.04–0.07 g/0.2–0.26 SQRT (g)) withdrawal thresholds than the naïve non-cannulated groups (p = 0.12–0.20). Following GTN injections, both the ICV (0.13–0.17 g/0.36–0.41 SQRT (g)) and naïve (0.17–0.33 g/0.41–0.58 SQRT (g)) mice presented with allodynia 2 hrs later on day 1 and 3 (p = 0.001 and p = 0.003, respectively, vehicle IP + GTN versus vehicle IP + vehicle). As reported previously (19,23), olcegepant IP inhibited the development of allodynia on both days (day 1: Mean difference 0.87 g/0.5 (SQRT (g), p < 0.001; day 3: Mean difference 0.57 g/0.45 SQRT (g), p = 0.001 olcegepant IP + GTN vs. vehicle IP + GTN); whereas olcegepant administrated ICV had no effect (day 1: mean difference −0.07 g/−0.1 (SQRT (g); day 3: mean difference 0.005 g/0.006 SQRT (g), p > 0.99 olcegepant ICV + GTN vs. vehicle ICV + GTN, both days). Taken together, these data strongly suggest that olcegepant is only effective in reversing GTN-induced allodynia when administered peripherally. This may indicate that the site of action of olcegepant, and hence CGRP receptor activation, is outside the blood-brain barrier.

Olcegepant prevents GTN-induced allodynia when given systemically but not intracerebroventricularly. The day after baseline measurements of 50% paw withdrawal thresholds, GTN (10 mg/kg IP) or vehicle was given just after olcegepant (1 mg/kg IP or 0.45 µg/mouse ICV) or vehicle on test day 1 and 3. The GTN response was measured 2 hrs after drug administration. Olcegepant IP clearly prevented the development of allodynia, whereas it was ineffective when given ICV. Raw data was square root transformed (SQRT) and presented as mean ± SEM.
The site of action of ALD405 is outside the blood-brain barrier
Following three days of rest after olcegepant administration, all mice were baseline tested again and allocated to new treatment groups. The next day, mice received ALD405 or the isotype control antibody either ICV or IP. One and 3 days later, the mice were administrated GTN to induce allodynia (Figure 3). The analysis revealed an overall difference between groups (F(4,37) = 24.01, p < 0.001). As seen previously, the cannulated mice had marginally lower baseline values (0.004–0.12 g/0.06–0.34 SQRT (g)) than the non-cannulated mice (p = 0.48–0.95) both before and after ALD405 delivery. IP administration of ALD405 prevented the development of allodynia both at 24 and 72 hours (24 hrs: Mean difference 1.0 g/0.69 SQRT (g); 72 hrs: Mean difference 0.84 g/0.61 SQRT (g), both time points p < 0.001 ALD405 IP + GTN vs. IgG control IP + GTN). In contrast, ICV administration had no effect at either timepoint (24 hrs: Mean difference 0.002 g/0.007 (SQRT (g); 72 hrs: Mean difference −0.01 g/−0.09 SQRT (g), both time points p > 0.99 ALD405 ICV + GTN vs. IgG control ICV + GTN). Albeit a minor fraction of antibody may penetrate to the brain after systemic administration (4,5), our results strongly suggest that the brain is unlikely to be the site of action of CGRP monoclonal antibodies.

ALD405 prevents GTN-induced allodynia when given systemically but not intracerebroventricularly. Baseline measurements of 50% paw withdrawal thresholds were obtained the day before and the day after dosing of ALD405 (10 mg/kg IP or 10 µg/mouse ICV). On test day 1 (24 hrs) and 3 (72 hrs) measurements were obtained 2 hrs after GTN (10 mg/kg IP). ALD405 IP clearly prevented the development of allodynia but was ineffective when given ICV. Raw data was square root transformed (SQRT) and presented as mean ± SEM.
ICV mice respond to olcegepant given intraperitoneally
Finally, we performed a positive control experiment in which the cannulated mice were administered olcegepant by IP injection (Figure 4). Exposure to olcegepant resulted in a highly significant response; the 50% withdrawal threshold of the olcegepant IP + GTN group was 0.49 g/0.7 ± 0.06 SQRT (g) versus 0.1 g/0.31 ± 0.07 SQRT (g) in the vehicle IP + GTN group (p < 0.001). This confirms that the lack of effect of the ICV injections was not caused by an intrinsic problem with cannulated mice but by the route of administration.
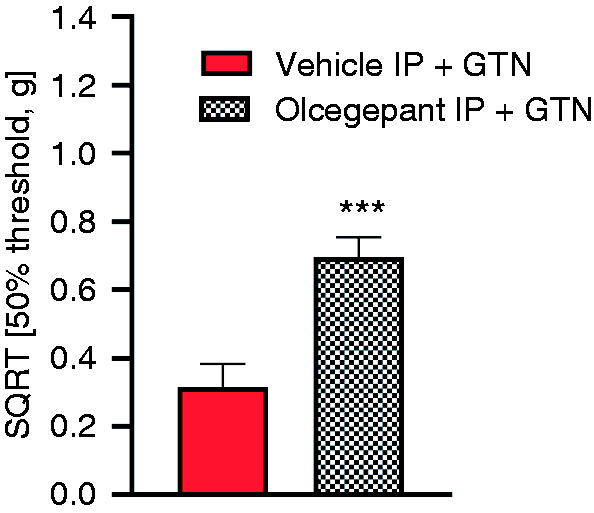
Positive control experiment: Olcegepant, IP prevents GTN-induced allodynia in ICV mice. Olcegepant (1 mg/kg IP) was given 10 minutes prior to GTN (10 mg/kg IP) and 50% withdrawal thresholds measured 2 hrs later. Raw data was square root transformed (SQRT) and presented as mean ± SEM.
Discussion
Data show that CGRP and its receptor are strongly involved in triggering migraine pain and pharmaceutical targeting of these signal molecules has been hailed to represent a new era in treatment (24). Regardless, the site of action has remained debated. Our study shows that neither a small molecule CGRP receptor antagonist nor a monoclonal CGRP neutralising antibody had efficacy given ICV in a validated mouse model of migraine pain. In contrast, both drugs were highly effective given systemically through IP administration. Collectively, this suggest that in GTN-induced migraine CGRP and its receptor are acting outside the brain with regards to the development of allodynia. Nitric oxide liberated from GTN freely passes the blood-brain barrier (25) and activates several brain areas relevant to migraine (26,27) and also produces migraine-associated symptoms other than pain (28,29). GTN likely causes CGRP release within the trigeminovascular system via TRPA1 activation (23,30), resulting in a general hyperexcitation of central pain circuits (31). Here, GTN-induced allodynia was measured through quantification of tactile sensitivity at the level of the hind paw, which has shown good correlation to cephalic allodynia (17,19,32–34). In humans, altered withdrawal reflexes of the lower limb are described following GTN treatment (35,36). From human studies, it is clear that GTN causes headache and no other pain (37) and because sumatriptan (known to alleviate migraine pain specifically (38–41)) is effective in the applied model (17,32) we argue that hind paw allodynia is a proxy for the presence of migraine pain and can be explained by spinal sensitization following activation of trigeminal pain circuits by GTN (35).
Administration of olcegepant and ALD405
A critical question for this study is whether the ICV doses used are sufficient for the drugs to be effective. The CSF-plasma ratio of the gepants is generally considered to be low (15,42) and was reported to be 2% for gepant MK-3207 (15), and is likely lower for olcegepant (R Hargreaves, personal communication). If olcegepant works via a central effect, 2% of the dose given peripherally must account for this effect. We aimed to inject a dose higher than 2% of the effective systemic dose of olcegepant to make sure we would not miss a possible central effect. We have previously shown the effect of olcegepant 1 mg/kg IP (19) and in-house unpublished data also show efficacy of 0.5 mg/kg IP. Given the poor oral bioavailability of olcegepant (42), it is unlikely that the entire dose given IP will reach the plasma. If 50% reaches the plasma, this equals 15 µg (of 1 mg/kg given to a 30 g mouse) of which the ICV dose of 0.45 µg equals 3%. Doing the same calculation for 0.5 mg/kg, we reach 6% of the active systemic dose. Thus, our ICV dose was higher than the fraction of olcegepant reaching the brain when used therapeutically. Importantly, a previous study showed that this dose administrated as 0.5 nmol olcegepant (resulting in 0.45 µg olcegepant HCl) effectively inhibited photophobia induced by CGRP also administered ICV in transgenic mice overexpressing RAMP1 (22). This supports that the selected dose was high as it prevented the effect of exogenous CGRP in mice overexpressing the receptor. Importantly, no adverse effects were reported (22).
ALD405 was given in a dose equal to 3.3% of the IP dose. This is beyond the fraction of antibody one would expect to enter the brain in the clinical setting. At peak concentrations, the monoclonal CGRP antibody galcanezumab was found in fractions of 0.12–0.34% of the plasma concentration in CSF, cerebellum, prefronatal cortex, spinal cord and hypothalamus of rats (5), supporting that our ICV dose of ALD405 was more than enough to be centrally effective.
We did not examine different time points of administration of ICV drugs in relation to GTN due to the multiple groups already in the present design. The ICV drugs were administrated at the same time as for IP dosing to provide enough time for the drugs to reach the brain parenchyma after delivery (43). Hence, if the chosen time points were adequate for IP deliveries to reach the brain (in case this was the site of action), it should also have been enough time for ICV drugs to reach the same site in the brain from the lateral ventricle. Contrarily, the relatively large ICV doses might have been actively transported out of the brain (44–46) to reach equilibrium at a lower dose (equal to what reaches the brain after IP delivery). In such a scenario, this smaller fraction might have been degraded before we performed behavioural testing resulting in a false negative result. However, it seems unlikely that this could have happened to both olcegepant and ALD405. In the previously mentioned study, where transgenic mice received both olcegepant and CGRP, ICV light sensitivity was measured already 30 min after the injection (22). However, this was in a migraine model based on CGRP and olcegepant both given ICV and thus quite different to the complex cascade of events behind the GTN model (23,47–49). In a setup more comparable to ours in complexity, the antinociceptive effect of lysine acetylsalicylate (aspirin) ICV was tested in the mouse formalin test and was found to be effective for up to 200 minutes (50). Collectively, these different data suggest that both the applied doses and timing of behavioural testing were relevant to test the efficacy of olcegepant and ALD405.
Study limitations
Our data were obtained in the GTN mouse model, which has been extensively used to model human migraine (18,19,51,52). However, it is a mouse model and its results cannot be translated to migraine patients with certainty. Nevertheless, the GTN model in both humans and rodents is well characterised in terms of its face and back-translational validity to migraine (28,29,52–54). Here, we did not study symptoms of migraine such as cortical spreading depression or light sensitivity thought to originate within the brain (55). Thus, our conclusions are restricted to migraine pain, no other symptoms.
Methodologically, limitations occur. Firstly, the control mice for the IP (systemic) administration of olcegepant and ALD405 were naïve and housed under different conditions. Ideally, they would have been from the same vendor, housed similarly and surgically prepared with ICV cannulas to avoid confounding bias from differences in basic mechanical sensitivity due to the implanted cannula and previous experiments. We did not include the extra 26 control ICV mice due to their high cost and for animal welfare reasons. However, comparisons were made within the naïve animals and within the ICV animals and, importantly, we distributed mice in test groups balanced with regards to their previous treatment. Thus, potential confounders were controlled for in the study design. Furthermore, we included a positive control for the ICV mice to demonstrate that the lack of effect of ICV olcegepant and ALD405 was not due to the ICV mice being non-responsive. Secondly, due to the relatively low number of ICV mice available, we did not include an ICV vehicle-vehicle negative control group. Again, the positive control experiment performed last confirmed that the sensitivity of the ICV mice was a GTN effect and not an effect of the implanted cannula. Third, the study was performed using male mice only. This is a limitation, since migraine in more prevalent in women (56). However, previous studies using both male and female animals report no differences between the sexes in the acute GTN response (17,18,33) and in this particular study we were worried that the smaller weight of female mice would make them more vulnerable to weight loss due to the ICV surgery. Thus, the experiments were carried out in males and we are fairly confident that a possible sex difference in blood-brain permeability would not have changed our conclusions, as males have been shown to have the more permeable blood-brain barrier of the two sexes (57).
Clinical implications
Our data suggest that CGRP released outside the brain is relevant in the GTN migraine model. One could argue that if the causative CGRP release of a true migraine resides within the brain, the present study is of no relevance. However, CGRP was not increased in blood from the internal jugular vein draining the brain during a migraine attack (58), while an increase in the external jugular vein draining extracerebral tissues has been described (59). The enhanced secretion of CGRP must originate from outside the blood–brain barrier (most likely the trigeminal ganglia) as the blood-brain barrier is not permeable to the CGRP peptide (60). The effect of small molecule CGRP receptor antagonists in the treatment of acute migraine attacks has generally been smaller than the effect of triptans (61,62). Similarly, the effect size of CGRP/CGRP receptor antibodies in migraine prophylaxis is only moderate, roughly 50% of patients have 50% or more reduction in attack frequency (63–71). One possibility for the modest effect of CGRP antagonism is that CGRP is not a dominating migraine mechanism (72,73). The other possibility is that migraine mechanisms are central; that is, in the brain. In that case, the minute quantities of CGRP receptor antagonists and CGRP antibodies that penetrate the blood-brain barrier might not be enough for full efficacy. There have been attempts to develop brain penetrant CGRP receptor antagonists (15), but no results are in the public domain. Should such efforts be intensified, or are the CGRP mechanisms of migraine extracerebral? Our results strongly support the latter. Taken together with other evidence such as migraine provocation by CGRP, which does not cross the blood-brain barrier in acute experiments (12), our results suggest that brain-penetrant CGRP antagonists will have no better efficacy than the non-penetrant substances available today.
Key findings
In the GTN mouse model of migraine pain, olcegepant and ALD405 are effective when given peripherally but not centrally. These CGRP-antagonising drugs elicit their effect on migraine pain outside the blood-brain barrier.
Footnotes
Author contributions
SLC, DMK and JO designed the study. SLC, CE and DMK performed all experiments and data analysis. SLC, DMK and JO wrote the manuscript. All authors read and approved the final manuscript.
Data availability
Raw data are available upon request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by Candys Foundation.