
Abstract
Background
Calcitonin gene-related peptide (CGRP) is a neuronal transmitter present in intracranial sensory nerves, where it is involved in migraine pathophysiology as well as other biological functions. Recently, the fully human monoclonal antibody erenumab (AMG 334), which targets the canonical calcitonin gene-related peptide receptor, showed significant prophylactic efficacy and favourable safety in phase II and III clinical trials for episodic and chronic migraine and is now approved for migraine prevention in several countries.
Objective
Given that calcitonin gene-related peptide can mediate vasodilation, we investigated the effect of erenumab on vasoactive responses in the presence or absence of various vasodilatory and vasocontractile mediators in a model using isolated human cerebral and meningeal arteries.
Methods
Ring segments of human isolated cerebral and meningeal arteries were mounted in a sensitive myograph. On arterial segments pre-contracted with 30 mM potassium chloride, vasoactive responses to calcitonin gene-related peptide were studied in the presence of different concentrations of erenumab. At the maximal tested inhibitory concentration of erenumab (100 nM), functional arterial relaxation in response to nicardipine or substance P, and the contractile responses to sumatriptan and dihydroergotamine were examined.
Results
30 mM potassium chloride produced a stable contraction of the vessel segments and calcitonin gene-related peptide induced a concentration-dependent relaxation. We observed that (i) erenumab had no direct contractile or relaxant effects per se (by itself), (ii) pre-treatment with erenumab antagonized the calcitonin gene-related peptide-induced relaxation in a competitive manner, (iii) the relaxant responses to nicardipine or substance P were unaffected in the presence of erenumab and (iv) the contraction induced by sumatriptan or dihydroergotamine was not modified by erenumab.
Conclusion
Our findings demonstrate that erenumab, while not associated with vasoactive properties per se, specifically inhibits calcitonin gene-related peptide-induced relaxation of cranial arteries without impacting vasodilatory responses or contractile responses of endogenous or pharmacological vasoactive compounds.
Keywords
Introduction
There is strong evidence for a role of calcitonin gene-related peptide (CGRP) and the calcitonin receptor-like receptor/receptor activity-modifying protein 1 (CLR/RAMP1, the CGRP receptor) in migraine pathophysiology where: (i) The trigeminovascular system has been suggested to be the migraine pathway, with CGRP and the CGRP receptor (CGRP-R) being expressed throughout this system (1,2); (ii) experimental stimulation of the trigeminovascular system results in increased levels of CGRP in cranial venous blood (3); (iii) jugular blood samples show that serum CGRP levels increase during migraine attacks (4,5); and (iv) studies blocking the CGRP-R or the CGRP ligand are effective in treating migraine pain (6). To date, the available prophylactic therapies have not been specifically developed to target the pathophysiology of migraine. Over the last years, several molecules that were developed to specifically target the trigeminovascular system and the associated CGRP-R have been released to the market. All these molecules have shown positive outcomes, illustrating the importance of CGRP in migraine pathophysiology (6).
Erenumab is a monoclonal antibody (mAb) that targets the CGRP-R with high potency and specificity and is therefore a different class of molecule from the other mAbs, which target the αCGRP ligand specifically. The CGRP ligand and CGRP-R mAbs are therefore expected to have different functional kinetics. Unlike CGRP ligand-binding mAbs, the ex vivo effect of erenumab on arteries can be studied in a myograph setup.
While βCGRP is mainly an enteric peptide, αCGRP is mainly expressed in sensory nerves (7). αCGRP is released from C-fibers that innervate the human vasculature, including the cerebral and meningeal arteries (8,9). The origin of a migraine attack is not yet fully understood, but most likely lies in the central nervous system (10). In contrast, the origin of pain is most likely peripheral and linked to CGRP release (6). This is supported by evidence of the mAbs' work outside the blood-brain barrier (BBB) (11,12). Although the specific site of activity is currently being discussed, the meningeal arteries and cerebral arteries are considered good proxies from the trigeminovascular system, where CGRP leads to vasodilation (13).
Since erenumab will bind the CGRP-R on arteries, this is a golden opportunity to calculate its pharmacodynamics on human tissue. Furthermore, as erenumab directly binds the CGRP-R on the arteries, it is interesting to examine whether it affects other functional responses. Therefore, our present work aimed to: (i) Study the effect of erenumab per se (by itself), (ii) investigate the antagonistic effects of erenumab on the vasorelaxation induced by αCGRP in human cerebral and meningeal arteries, and (iii) observe any possible effect of erenumab on other vasoconstrictors and vasodilators.
Methods
Human isolated arteries
Human cerebral cortex arteries (four males, two females, age 43–75 years) and middle meningeal arteries (two males, three females, age 48–83 years) with internal diameters of 300–500 µm, were removed in routine neurosurgical tumor-removal operations in Lund, Sweden. All vessels were placed in physiological buffer solution with a composition in mM of: NaCl, 119; KCl, 4.7; CaCl2, 1.5; MgSO4, 1.17; NaHCO3, 25; KH2PO4, 1.18; EDTA, 0.027; glucose, 5.5, pH 7.4, aerated with 5% CO2 in O2 (carbogen) and transported to the laboratory. The study was approved by Lund University Ethics Committee (LU99-818-01) and all patients gave informed consent.
Functional experiments
The arteries were cut into cylindrical segments of 1 or 2 mm in length for ex vivo pharmacological experiments. Using this procedure there will be some nesting of the data, since patient data are used for several concentrations of the mAbs. Due to the relatively low sample size on human samples, we did not model any nesting effect. Each segment was mounted on two metal prongs, one of which was connected to a force displacement transducer, and vascular tension was increased by adjusting the distance between the metal prongs (13,14). The arteries were maintained in physiological buffer solution continuously gassed with carbogen (37℃) and allowed to equilibrate for approximately 30 minutes. The vessel tension was continuously recorded and the distance between the pins or wires was adjusted to maintain a resting tone of 4 mN for cerebral and meningeal arteries or stretched to a tension normalized to 90% of l100 (the diameter when transmural pressure equals 100 mmHg (14)) for the meningeal segments. Contractile capacity of each arterial segment was examined by exposure to a 60 mM K+ physiological buffer (normal physiological buffer where NaCl had been exchanged on an equimolar basis for KCl).
The per se effect of erenumab or the IgG control was tested on arterial segments at baseline or precontracted with 30 mM KCl. The force produced was observed for around 30 minutes following the addition of control IgG or erenumab.
The relaxant effect of human αCGRP was examined by means of a cumulative concentration response curve, in the absence or presence of various concentrations of erenumab (1 nM, 10 nM and 100 nM). Relaxation curves to αCGRP (10 pM–100 nM) were performed on arteries precontracted with 30 mM KCl. Similarly, relaxation curves to nicardipine (100 pM–10 µM) was performed on arteries precontracted with 30 mM KCl in the absence or presence of erenumab (100 nM). Furthermore, cumulative concentration-response curves in the absence or presence of the antagonist erenumab (100 nM) were performed for sumatriptan (1nM–10 µM) and dihydroergotamine (DHE, 100 pM–1 µM). The functional integrity of the endothelium was determined in the absence or presence of erenumab (100 nM) by observing relaxation to a single concentration of substance P (SP, 10 nM) on arteries pre-contracted with U46619 (100 nM).
Compounds
The following materials were used in the ex vivo experiments: Human αCGRP (NeoMPS S.A., Strasbourg, France, and Sigma, USA), SP, sumatriptan, dihydroergotamine, and nicardipine (Sigma Aldrich, Germany). Erenumab (AMG334) or its IgG control were obtained from Amgen (CA, USA). The agonists were dissolved in saline and stored as aliquots at −20℃. Erenumab was delivered in a stock solution, stored at −20℃, and further diluted in saline when used.
Analysis of data
The vasodilator response to αCGRP was expressed relative to the contraction evoked by 30 mM K+ buffer (=100%). The constrictive response to sumatriptan and DHE were expressed in relation to the contraction elicited by 60 mM K+ buffer solution. For each segment, the maximum vasodilator or vasoconstrictor effect (Emax) was calculated. The concentration-response curves for all agonists were analysed with nonlinear regression analysis using a four-parameter concentration-response curve fit, which also fits the Hill Slope from the data (Y = Bottom + (Top-Bottom)/(1 + 10^((logEC50−X)*HillSlope)). The potency of agonists was expressed as logEC50 (i.e. the logarithm of the molar concentration of agonist inducing half maximum response) using GraphPad Prism 8.0.2 (GraphPad Software Inc., San Diego, CA, USA). To analyse the potency of erenumab, a Schild plot was generated for the cerebral arteries and an apparent pKB was calculated for the meningeal artery. Data are expressed as mean values ± SEM and ‘n’ refers to the number of patients from whom the arteries were collected. Statistical difference was calculated by two-way ANOVA with Sidak's multiple comparison. The logEC50 values were compared using one-way ANOVA with Dunnet's multiple comparison.
Results
Human cerebral (cortical) and meningeal arteries were removed from macroscopically normal tissue during neurosurgical operations for intracranial tumors. After two repeated 60 mM KCl-induced contractions followed by washout, 30 mM potassium buffer was applied to elicit a stable pre-tension where the endothelial function was evaluated by application of one concentration of SP (10 nM). The vessels relaxed by 51 ± 7%, which compared well to other studies of human vasculature (14).
Effects of erenumab on functional relaxations to αCGRP in human cerebral and meningeal arteries
The CGRP-R mAb erenumab or its IgG control did not show any vasomotor responses per se in any of the isolated artery segments at basal tone or precontracted with 30 mM KCl (Figure 1). In the meningeal artery, αCGRP induced a concentration-dependent relaxation of 30 mM K+ in the control (Emax = 63 ± 16%, logEC50 = −7.2 ± 1.9), and in the presence of 1 nM (Emax = 51 ± 27%, logEC50 = −8.1 ± 0.4), 10 nM (Emax = 65 ± 21%, logEC50 = −7.6 ± 1.0) and 100 nM (Emax = 32 ± 11%, logEC50 = −6.7 ± 0.1). There was a significant inhibition of αCGRP-induced relaxations at 100 nM of erenumab at 3 · 10−7 M CGRP (p = 0.0391, Figure 2(a)). In the cerebral arteries (Figure 2(b)), we observe a more potent dilation to αCGRP, which gave more evident shifts in the response to αCGRP. The control had an Emax of 93 ± 17% and a potency of logEC50 of −9.5 ± 0.5, which was weakly attenuated by 1 nM (Emax = 102 ± 17%, logEC50 = −9.2 ± 1.9), 10 nM (Emax = 79 ± 13%, logEC50 = −8.3 ± 0.3) and significantly attenuated by 100 nM erenumab at 3 · 10−9 M (p = 0.0319) and 10−8 M CGRP (p = 0.0449) and caused a significant shift in the logEC50 value for CGRP (Emax = 42 ± 3%, logEC50 = −7.0 ± 1.2, pEC50shift = 0.0465). The concentration-effect antagonism by erenumab in the cerebral arteries allowed us to perform a Schild plot analysis (Figure 2(c)). The slope was not significantly different from 1 (1.22 ± 0.25), indicating competitive inhibition. The potency, calculated as the pA2 value, was 9.00 (10.44–8.57, 95% confidence interval). For the meningeal artery, the current data did not allow Schild plot analysis, so instead we calculated the apparent pKB, which was 7.50 (7.08–7.83, SEM interval). For a competitive antagonist (i.e. one where the slope of the Schild plot equals 1), the pKB is theoretically equal to the pA2 value. Hence, it appears that the concentrations needed to inhibit CGRP-induced vasodilation in meningeal arteries are larger than for cerebral arteries.
Sample trace for the per se effect of erenumab. (a) Cerebral artery pre-contracted with 30 mM K+ exposed to different concentrations of erenumab or control. αCGRP was later added as a concentration-response curve. (b) Cerebral artery incubated with different concentrations of erenumab or control. Afterwards, 10−6 M U46619 was added to induce contraction. Relaxant effect of αCGRP on human intracranial arteries. (a) meningeal and (b) cerebral arteries, which were precontracted with 30 mM KCl. Concentration response curves to αCGRP in the absence or presence of increasing concentrations of erenumab. There was a clear shift to the right in the concentration effect curves. (c) Calculated Schild plot for the cerebral arteries. Values given represent mean ± SEM, number of subjects = 5–6.

Other vasoactive agents
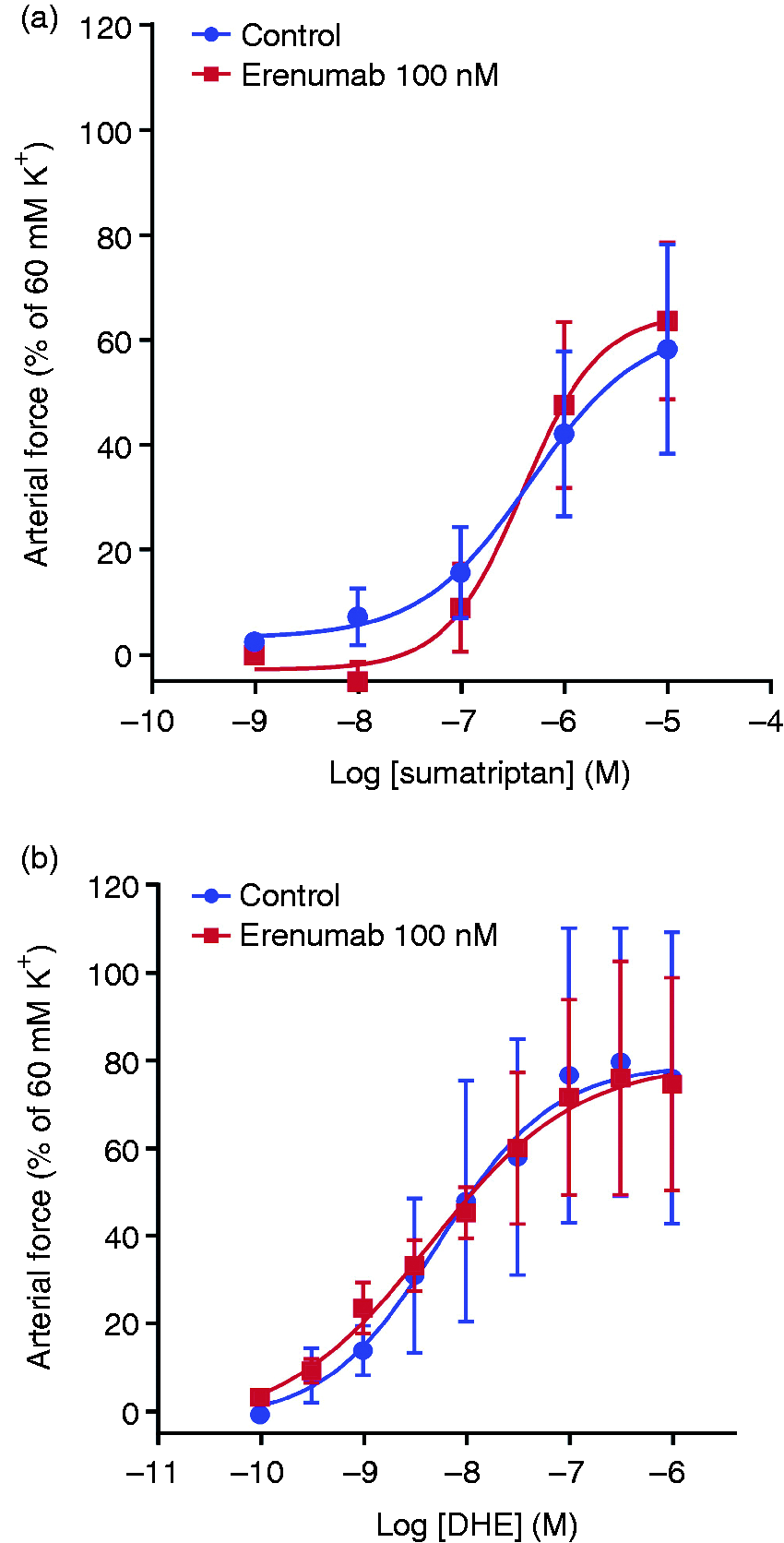
At a concentration of erenumab that was high enough to significantly block the αCGRP induced relaxation (functional blockage, 100 nM), we investigated other vasoactive compounds in intracranial arteries. We combined meningeal and cerebral arteries, as there was no difference in the effect of the compounds tested (Supplemental Figure 1). The calcium ion-channel blocker nicardipine elicited a concentration-dependent relaxation of precontracted cerebral arteries (Emax = 83 ± 10%, logEC50 = −8.0 ± 0.5) in the presence of IgG control), which was not altered by the presence of erenumab (Emax = 91 ± 6%, logEC50 = −7.7 ± 0.2) (Figure 3). The relaxant response to SP remained at levels similar to before the test with erenumab (control IgG = 51.9 ± 28% versus 100 nM erenumab = 49.9 ± 24%). The two acute migraine drugs sumatriptan (Emax = 58 ± 20%, logEC50 = −6.4 ± 0.3) and DHE (Emax = 78 ± 29%, logEC50 = −8.3 ± 0.6) elicited concentration-dependent contractions when applied to relaxed vessel segments (Figure 4). The presence or absence of erenumab or its IgG control did not alter these concentration-response relationships for sumatriptan (Emax = 64 ± 15%, logEC50 = −6.3 ± 0.7) or DHE (Emax = 79 ± 22%, logEC50 = −8.4 ± 0.7).
Relaxant effect of nicarpidine on human intracranial arteries that were precontracted with 30 mM KCl with or without 100 nM erenumab. Values given represent mean ± SEM, number of subjects = 5. Contractile responses to sumatriptan (a) or DHE (b) with or without 100 nM erenumab on intracranial arteries. Values given represent mean ± SEM, number of subjects = 5.
Discussion
Studies of CGRP-R were long hampered by (i) lack of understanding of the receptor construction and (ii) the lack of a specific blocker. By identifying the RAMPs as necessary components, McLatchie et al. showed that the CGRP-R consists of two subunits, the CLR and RAMP1 (15). The lack of a specific blocker was solved by producing the small molecule competitive CGRP-R antagonists, a class called gepants, which were found to have good efficacy in acute treatment of migraine attacks (16,17). The mAbs targeting the CGRP ligand or the CGRP-R have shown efficacy as prophylactics in episodic and chronic migraine (18,19). Erenumab is one such mAb, which targets the CGRP-R specifically by binding to the N-terminals of CLR and RAMP1. As CGRP has a direct vasodilatory effect, part of erenumab's effect could be caused by preventing vasodilation. Furthermore, as erenumab binds directly to CGRP-Rs on the arteries, any potential effects on other vasoactive agents are important to investigate.
Here we have studied the pharmacological inhibition on αCGRP combined with calculation of potency on human tissue. Erenumab had no effect per se (by itself), but caused an antagonism of αCGRP-induced vessel relaxations that shifted the logEC50 (Figure 2). We cannot reach a conclusion regarding the shift in Emax, as we did not reach a plateau. Erenumab binding is believed to be fully reversible and potentially fully displaceable by CGRP, hence we would not expect any effect on Emax. This is also confirmed with the slope not being significantly different from 1 (Figure 2). At a concentration that significantly inhibited specific αCGRP-induced relaxations, erenumab did not modify the relaxation to nicardipine (which dilates arteries through inhibition of Ca2+ channels, Figure 3) or SP, nor the contractions induced by DHE or sumatriptan (Figure 4). Similar findings have been reported for the small molecule CGRP-R blocker telcagepant (14). Thus, from a pharmacological point of view, erenumab influenced CGRP-induced dilation but did not alter any of the other vasomotor responses examined. Therefore, blocking the CGRP-R with erenumab may not be theoretically associated with potentially harmful direct vasoconstriction of cerebral and meningeal blood vessels.
Available observations in vivo suggest that CGRP-Rs normally do not have a tonus role (5). Therefore, the profile of erenumab agrees well with previous studies of CGRP-R antagonists such as CGRP8-37 (20), olcegepant (21–23) and telcagepant (14). In comparing the clinical effects of erenumab to the present data, our concentration dependence is matching the clinical efficacy at 66 nM (24). Furthermore, clinically, the mean serum steady-state true concentration of erenumab was 6.1 ± 2.1 µg/mL (∼40 nM) and 15.8 ± 4.8 µg/mL (∼105 nM) for the 70 mg and 140 mg doses, respectively (25), corresponding well to the in vitro effects we observed. We found an ex vivo pA2 value in this study of 1 nM (Figure 2) for the cerebral artery and of around 30 nM for the meningeal artery, and significant shifts in the logEC50 and relaxation in response to αCGRP at 100 nM. Since the concentration of drug equal to the pA2/pKB value may not be sufficient to decrease functional responses (since it only shifts the concentration-response curves two-fold to the right), this might explain why some patients will need the 140 mg dose for effect.
Since penetration of erenumab through the blood-brain barrier (BBB) is unlikely to achieve anti-migraine efficacy due to its high molecular weight (12,26), the most likely target is outside the BBB. There are, in our opinion, three potential targets: i) The meningeal vasculature (shown in the current study); ii) peripheral synapses of Aδ neurons; or iii) the soma of Aδ neurons in the trigeminal ganglion (TG). There is contrasting evidence for an involvement of vasodilation in migraineurs (27,28), and most likely it is an epiphenomenon from the increased CGRP release. Nevertheless, erenumab does inhibit functional dilation by αCGRP in both meningeal and cortical arteries. The two other potential targets include the nociceptive Aδ neurons (29). This is supported by a study showing that pretreatment in rodents with the CGRP ligand mAb fremanezumab selectively inhibited the responsiveness of trigeminal Aδ neurons, but not C-fiber neurons in rats, suggesting that Aδ meningeal nociceptor neurons are likely targets for prevention of headache (30). The CGRP-R is strongly expressed on the soma of the trigeminal neurons within the TG (31) and they can be functionally activated by CGRP (32). Furthermore, the TG also resides outside the BBB (11), making it a possible target. Further studies are needed to resolve the key question: Where is the definite site of action for the therapeutic mechanisms involving the CGRP-R?
Conclusion and clinical perspective
In conclusion, erenumab antagonizes relaxations induced by CGRP with a potency similar to the clinical potency, without affecting the vascular tone per se. In the current study, we show that erenumab does not affect a vasodilator (nicardipine, a drug used to treat high blood pressure and angina pectoris) or SP with a different mode of action. Furthermore, erenumab does not affect the vasoconstrictive properties of sumatriptan and DHE, which can then still exert their effects in the presence of erenumab. Erenumab and sumatriptan/DHE could therefore give potential additive beneficial effect. Although our study conclusively shows that erenumab does block vasodilation in response to αCGRP, it remains to be demonstrated whether inhibition of vasodilatation by αCGRP mediates the therapeutic action of erenumab, or if the target resides in other parts of the trigeminovascular system.
Supplemental Material
Supplemental material for Erenumab (AMG 334), a monoclonal antagonist antibody against the canonical CGRP receptor, does not impair vasodilatory or contractile responses to other vasoactive agents in human isolated cranial arteries
Supplemental Material for Erenumab (AMG 334), a monoclonal antagonist antibody against the canonical CGRP receptor, does not impair vasodilatory or contractile responses to other vasoactive agents in human isolated cranial arteries by Lena Ohlsson, Kristian A Haanes, Erik Kronvall, Cen Xu, Josefin Snellman and Lars Edvinsson in Cephalalgia
Footnotes
Key findings
Erenumab, while not associated with vasoactive properties per se, specifically inhibits CGRP-induced relaxation of cranial arteries.
Relaxant/contractile responses of other compounds were unaffected in the presence of erenumab.
The data show that erenumab is not expected to interfere with other vascular signaling pathways.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: LE has given lectures on CGRP for Amgen, Novartis, and Teva, and has received minor grant support (unrestricted grants) from Amgen/Novartis. CX is an employee (with stocks) of Amgen. JS is an employee (with stocks) of Novartis. LO, KAH and EK declare no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Swedish Heart-Lung Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.