
Abstract
Background
The current understanding of mechanisms behind migraine pain has been greatly enhanced with the recent therapies targeting calcitonin gene-related peptide and its receptor. The clinical efficacy of calcitonin gene-related peptide-blocking drugs indicates that, at least in a considerable proportion of patients, calcitonin gene-related peptide is a key molecule in migraine pain. There are several receptors and molecular pathways that can affect the release of and response to calcitonin gene-related peptide. One of these could be purinergic receptors that are involved in nociception, but these are greatly understudied with respect to migraine.
Objective
We aimed to explore purinergic receptors as potential anti-migraine targets.
Methods
We used the human middle meningeal artery as a proxy for the trigeminal system to screen for possible anti-migraine candidates. The human findings were followed by intravital microscopy and calcitonin gene-related peptide release measurements in rodents.
Results
We show that the purinergic P2Y13 receptor fulfills all the features of a potential anti-migraine target. The P2Y13 receptor is expressed in both the human trigeminal ganglion and middle meningeal artery and activation of this receptor causes: a) middle meningeal artery contraction in vitro; b) reduced dural artery dilation following periarterial electrical stimulation in vivo and c) a reduction of CGRP release from both the dura and the trigeminal ganglion in situ. Furthermore, we show that P2X3 receptor activation of the trigeminal ganglion causes calcitonin gene-related peptide release and middle meningeal artery dilation.
Conclusion
Both an agonist directed at the P2Y13 receptor and an antagonist of the P2X3 receptor seem to be viable potential anti-migraine therapies.
Introduction
The therapies for migraine patients have recently made a huge step forward with the arrival of the monoclonal antibodies targeting calcitonin gene-related peptide (CGRP) or its receptor (1). Apart from directly scavenging CGRP or blocking its receptor, future therapies could also be directed at regulating the cellular effect of CGRP or the neuronal release of CGRP or other neuropeptides. Currently available, acutely acting antimigraine drugs, the triptans, have also been demonstrated to inhibit CGRP release (2), which could be one of their most important therapeutic actions.
Neither the origin of a migraine attack, nor the cause of migraine pain is fully understood. In contrast, the involvement of the CGRP-ergic system in migraine has been clearly recognized (1). For example, CGRP is released during a migraine attack, and mitigation of the migraine pain after the use of triptans is accompanied by a normalization of CGRP levels (3). Hence, molecular targets that can prevent CGRP release or counteract the effects of CGRP are viable targets for anti-migraine therapy, as exemplified by the fact that all CGRP-targeted therapies thus far have been successful (4).
An alternative approach to affect the CGRP-ergic system may be targeting the purinergic receptors because: i) purinergic receptors are involved in pain, ii) a subgroup of purinergic receptors initiates similar signaling pathways to the triptans and iii) in other systems, purinergic receptors have been shown to modulate neurotransmitter release. There are, in all, 19 purinergic receptors known today (18 in rodents), and there is increasing evidence for the involvement of these receptors as modulators or tuners of cellular responses (5). Purinergic receptors are divided into two main groups: The adenine nucleoside receptors (A/P1 receptors) and the purine/pyrimidine nucleotide receptors (P2 receptors). Furthermore, the P2 receptors are divided into P2X receptors (P2X1-7), which are ligand-gated ion channels, and P2Y receptors (P2Y1,2,4,6,12-14), which are G protein coupled receptors (5). The purinergic field is relatively new, which is exemplified by the presence of only one marketed drug that targets purinergic receptors, namely the anti-coagulant clopidogrel (6).
Purinergic research in migraine has mainly been focusing on ATP and the role in neuronal sensitisation by acting on the low threshold ligand gated P2X3 receptor ion channel (7). However, there is also data on the high ATP threshold P2X7 receptor in migraine models (8). Focusing on the P2X3 receptor, increased expression in trigeminal ganglion (TG) neurons was observed in vitro after exposure to CGRP and in vivo following induction of trigeminal-associated pain (9,10). Activation of the P2X receptors might explain the painful sensation of ATP (11). In contrast, the more modulatory P2Y receptors have been given little attention and to our knowledge only one in vitro study, investigating calcium signalling in primary neuron-glial trigeminal cultures, exists on the role of P2Y receptors (12).
Activation of three purinergic receptors, the P2Y12, P2Y13 and P2Y14 receptors, leads to a decrease in intracellular cAMP (13), as these receptors are coupled to Gi proteins. This is similar to the activation of 5-HT1B/1D receptors, which are the pharmacological target of the triptans (14). Activation of the 5-HT1B/1D receptors leads to vasoconstriction of the middle meningeal artery (MMA) and reduces the release of CGRP from peripheral C-fibers (15). In contrast to the 5-HT1B/1D receptors, with an abundance of data available, there is limited evidence for the negative modulation of presynaptic transmission of sympathetic (16) and cholinergic nerves (17) following ADP-induced activation of the P2Y12 or P2Y13 receptors. It is not clear yet whether a similar action could exist for trigeminal nerves, which would have implications for the release of CGRP. We therefore embarked on a study with the aim of uncovering purinergic targets in line with the neurovascular theory of migraine.
We hypothesized that P2Y receptors could play an important modulatory role in the pathophysiology of migraine and that P2X receptors might be important in the initiation of a migraine attack. Using the human MMA (hMMA), a potential direct target for antimigraine medication, as well as a proxy for the trigeminal system, combined with intravital microscopy and CGRP release in rodents, we studied both a direct vasoactive effect, as well as a modulatory effect on CGRP release, by purinergic receptor stimulation in the trigeminal system.
Methods
All studies on animals were performed between 09.00 and 18.00, either on explanted organs or in the laboratory under anaesthesia (see below).
Myograph studies in hMMA
Samples of dura mater were perioperatively obtained from 10 patients (two males and eight females; 48–69 years) undergoing neurosurgical procedures. Tissues were collected in a sterile organ-protecting solution and were immediately brought to the laboratory. Subsequently, hMMAs (internal diameter 0.5–1.5 mm) were dissected and used the same day or stored overnight in cold oxygenated Krebs solution of the following composition: 119 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3 and 11.1 mM glucose; pH 7.4. Study protocols were approved by the ethics committee at the Erasmus Medical Center, Rotterdam.
The hMMAs were cut into 1–2 mm long segments and mounted on stainless-steel wires (Ø 40 µm) in Mulvany myographs (Danish Myo Technology, Aarhus, Denmark) filled with carbogenated Krebs buffer solution at 37 ℃. The segments were stretched to their optimal lumen diameter (0.9 × L100), where L100 is an estimate of the diameter of the vessel under a passive transmural pressure of 100 mmHg (13.3 kPa). Data were obtained through a LabChart data acquisition system (AD Instruments Ltd, Oxford, UK). hMMAs were exposed to 30 mM KCl, followed by 100 mM KCl to determine the reference contractile response. Using the indicated agonists, a cumulative concentration response curve (using half log-steps unless otherwise indicated) was constructed. This cumulative approach might lead to an underestimation of the maximal contractions in case of desensitization, but in view of the scarcity of human tissue an alternative approach, using only one single concentration per vessel segment, is not feasible. The functional integrity of the endothelium was tested with substance P (10 nM) on arteries precontracted with the thromboxane A2 analogue U46619 (100 nM). The same precontraction was used to construct vasodilation curves.
Intravital microscopy on a closed cranial window in rats
Twenty-seven normotensive male Sprague-Dawley rats (300–400 g), purchased from Harlan (Horst, The Netherlands), were maintained on a 12/12-h light-dark cycle (with light beginning at 7 a.m.) and housed at a constant temperature (22 ± 2 ℃) and humidity (50%), with food and water ad libitum. The animals were anaesthetized with sodium pentobarbital (60 mg/kg i.p., followed by 18 mg/kg i.v. per hour when necessary). The adequacy of anaesthesia was judged by the absence of ocular reflexes and a negative tail flick test. The Institutional Ethics Committee approved all experimental protocols (Erasmus MC; permission protocol number EMC 1931 [118-09-04]).
The protocol was identical to our previous studies (18). Briefly, the left femoral vein and artery were cannulated for intravenous (i.v.) administration of drugs and continuous monitoring of blood pressure. Rats were placed in a stereotaxic frame and the skull was carefully drilled thin, until the MMA was visible. An intravital microscope (Leica MZ 16; Leica Microsystem Ltd, Heerbrugg, Switzerland) was used to record the artery diameter with a cyan filter on a cold source of light. A zoom lens (80 × magnification) and camera (DCx V3.52, Thorlabs LTD, Ely, UK) were used to capture the image of the dural artery, which was displayed and measured on a computer using a dedicated software package (IDA-Intravital Dimension Analyser; http://www.beneryx.co.uk) integrated with an ADC/DAC board (DI-158, DATAQ instruments, ‘s-Hertogenbosch, The Netherlands). For the periarterial electrical stimulation, a bipolar stimulating electrode (NE 200X, Clark Electromedical, Edenbridge, Kent, U.K.) was placed within 200 µm from the MMA. The cranial window surface was stimulated at 5 Hz, 1 ms for 10 s (stimulator model S88, Grass Instruments, West Warwick, RI, USA). As we previously demonstrated, dural vasodilator responses are reproducible after repeated stimuli for at least four times (18,19). Animals were randomized, and we allowed 30 min between stimulations/treatments for recovery to the baseline diameter. MRS2211 (P2Y13 receptor antagonist) or ADPβS (ADP analogue, P2Y1/12/13 receptor agonist) was administered 15 min before periarterial electrical stimulation. The duration of each experiment was approximately 2.5 hours after stabilization. The detection limit for arterial diameter was about 10 µm; in case an artery was not visible due to pronounced constriction, its diameter was set to be 10 µm.
CGRP release
Twelve male Sprague-Dawley rats (300–400 g), purchased from Taconic (Ejby, Denmark) were maintained on a 12/12-h light-dark cycle (with dark beginning at 7 am) and housed at a constant temperature (22 ± 2 ℃) and humidity (55 ± 10%), with food and water ad libitum. Rats were generally housed in Eurostandard cages (Type VI with 123-Lid) 2–6 together and single housed (Type III with 123-Lid) directly before undergoing the procedure.
Rats were anaesthetized by CO2 inhalation and decapitated. All procedures are approved by the Danish Animal Experimentation Inspectorate. The protocol is described in detail elsewhere (19). Shortly, the trigeminal nucleus caudalis (TNC) was excised from the brain stem, measuring approximately 6 mm in length. The skull was cut mid-sagittally and the brain halves were carefully removed while the cranial dura was left attached to the skull, and the trigeminal ganglion (TG) was dissected out. TGs and TNCs were immersed in 10 ml synthetic interstitial fluid (SIF, composition: 108 mM NaCl, 3.5 mM KCl, 3.5 mM MgSO4 , 26 mM NaHCO3, NaH2PO4, 1.5 mM CaCl2, 9.6 mM NaGluconate, 5.6 mM glucose and 7.6 mM sucrose; pH 7.4.) at 37 ℃ for 30 min. Skull halves were transferred to a beaker containing SIF and washed two times (each wash, 15 minutes) in 250 ml SIF.
TGs and TNCs were randomized, placed in Eppendorf tubes in a heating block at 37 ℃. TGs and TNCs were again washed five times (each wash, 10 minutes), with 300 µl SIF. Both skull halves were randomized and placed in a humid chamber above a water bath to maintain temperature at 37 ℃ and washed five times (each wash 10 minutes) with 300 µl SIF. After 10 min incubation with 300 µl SIF, 200 µl samples for measuring the basal CGRP release were collected from all the tissues, mixed with 50 µl enzyme immunoassay buffer (containing protease inhibitors) and stored at −20 ℃ until analysis, within a week after the experiment was performed. Previous studies have shown that there is no significant difference between the basal CGRP release from the left and right side of the tissues; thus, one side served as a control for the other, which has been shown to reduce the experimental and biological variations (19). The release of CGRP from the skull halves, TG and TNC was induced by 60 mM potassium. To maintain equal osmolarity, a proportional amount of Na+ was removed from the buffer. Experiments by others have shown that 10 min incubation is sufficient for a significant and reproducible release of CGRP over basal levels (19). The tissues were pre-incubated for 20 minutes with different concentrations of inhibitors/vehicle and agonist/vehicle, and during the final KCl challenge the same concentrations were maintained.
The samples were processed using commercial EIA kits (SPIbio, Paris, France) to study CGRP release. From all tissues, 200 µl of sample was mixed with 50 µl of EIA buffer, which was also added to the CGRP standard. Determination of CGRP content was calculated based on this standard curve. Previous studies on the samples obtained from TNC showed that the amount of CGRP release was too high to fall in the linear range of the standard curve (19). Therefore, TNC samples were diluted six times. Antibody in the CGRP EIA kit is directed against human-CGRP α/β, but it has 100% cross-reactivity with rat and mouse CGRP (19,20). The protocol was performed following the manufacturer's instructions. Briefly, samples were incubated at 4 ℃ for 16–20 h, washed, and incubated with Ellman's reagent. Following 60 min of incubation, the optical density was measured at 410 nm using a micro-plate photometer (Tecan, Infinite M200, software SW Magellan v.6.3, Männedorf, Switzerland). Samples (a total of two dura and one TNC) that did not generate any CGRP release in response to 60 mM potassium, and one TG with baseline over 100 pg/ml CGRP, were not included in the analysis. Since data were paired, both the left and right side were excluded in the above cases.
Immunohistochemistry
Human TGs were collected at autopsy, within 48 h post-mortem, from four patients (two males and two females; 68–82 years). The patients were without disorders related to the central nervous system. The specimens were fixed after removal in 2% paraformaldehyde (PFA) and 0.2% picric acid in 0.1 mol/l phosphate buffer, pH 7.2, tyrode solution. The tissue samples were kept at −80 ℃ until embedding and cryo-sectioning. The study followed the guidelines of the European Communities Council (86/609/ECC) in accordance with the Szeged University Medical School guidelines for ethics in human tissue experiments and was approved by the local Hungarian Ethics Committee. The hMMA was obtained from the same patients that were used for the myograph studies. Study protocols were approved by the ethics committee at the Erasmus Medical Center, Rotterdam.
Human TGs and MMAs were cryosectioned. Details on immunohistochemistry are described elsewhere (21). In short, slides with 10 µm sections were washed in PBS containing 2.5% Triton (PBS-T) for 15 min. Next, the primary antibody, rabbit anti-P2Y13 receptor (Alomone, APR#017, 1:50), rabbit anti-P2X3 receptor (Alomone, APR#026, 1:200) or mouse anti-CGRP (Abcam Ab81887, 1:100) was added. Following incubation overnight in moisturized chambers (+8 ℃), cryosections were washed in PBS-T twice for 15 min and incubated for 1 hour with secondary antibodies (FITC anti-mouse, Jackson Immunoresearch 1:200, and Cy3 anti-rabbit, Jackson Immunoresearch, 1:300) in the dark. After three washes for 15 min, the slides were mounted with Vectashield mounting medium containing DAPI (4′,6-diamidino-2-phenylindole, Vector Laboratories, Burlingame, CA, USA). Negative controls were performed omitting the primary antibody. Immunofluorescence was observed using an epifluorescence microscope (Nikon 80i, Tokyo, Japan) combined with a Nikon DS-2MV camera. The images were then processed using Adobe Photoshop CS3 (v10.0 Adobe 3 Systems, Mountain View, CA, USA). Potential colocalization was determined by superimposing the images taken with different wavelength filters.
Data presentation and statistical evaluation
All quantitative data are presented as mean ± SEM. The n-number represents one rat or one human. The peak increases in dural meningeal artery diameter, as well as changes in mean arterial blood pressure (MAP, in vivo experiment) were expressed as percent change from baseline. The difference between two concentration response curves was determined by two-way analysis of variance (ANOVA) and the Bonferroni post-test. The difference between the variables within one group was compared by using a one-way repeated measures ANOVA followed by Dunnet's test. When only two variables were compared, a paired Student's t-test was used.
Compounds
The compounds used in this study were: Sodium pentobarbital (Nembutal; Ceva Sante Animale B.V., Maassluis, The Netherlands); MRS2211, αβmetATP, olcegepant (Tocris); ADPβS, UDPβS, UTPγS, ATPγS (Biolog.de) and CGRP (NeoMPS S.A., Strasbourg, France). All other chemicals were from Sigma Chemicals Co. (Steinheim, Germany). All compounds were dissolved in distilled water, with the exception of capsaicin and olcegepant, which were dissolved in DMSO. All solutions were further diluted in saline for in vivo studies or distilled water for in vitro studies.
Results
The MMA is an important component of the trigeminovascular system and may serve as a proxy for the neurovascular system and is therefore a good indicator of potential anti-migraine therapies, also those acting primarily at neuronal components of the trigeminovascular system. We have previously fully characterized the contractility of the rat MMA (rMMA) to a number of purinergic agonists and therefore initially set out to compare these data to the hMMA.
Myograph studies on the hMMA
hMMAs were obtained from 10 patients (two males and eight females; 48–69 years) during neurological procedures requiring trepanation of the skull.
In vitro relaxation of hMMA
Since the hMMA has a much larger diameter than the rMMA it can therefore be mounted in myographs without damaging the endothelium, making it possible to investigate endothelium-dependent vasodilation. The presence of functional endothelium was checked with the addition of substance P (79 ± 8% relaxation from 100 nM U46619) in the vessels used below. We investigated the main known purinergic vasodilators, UTPγS (P2Y2 and P2Y4 receptor agonist), UDPβS (P2Y6 receptor agonist) and ADPβS (P2Y1, P2Y12, and P2Y13 receptor agonist). ADPβS (Figure 1(a), pEC50 6.82 ± 0.28, EMAX 72 ± 10%) had similar potency as UTPγS (Figure 1(b), pEC50 6.27 ± 0.39, 84 ± 9%). UDPβS did not cause any vasodilation, but further contracted the artery by 45 ± 21% (Figure 1(c), pEC50 ∼ 4).
Purinergic-induced relaxations of hMMA with endothelium. Concentration-dependent relaxation induced by UTPγS (a), ADPβS (b), and UDPβS (c) on hMMAs precontracted with 100 nM U46619. The precontraction is set to be 100% above the baseline. Data are shown as mean ± SEM, n = 5.
In vitro contraction of the hMMA
For the contractile studies, we performed the experiment in endothelium-free hMMA, which was confirmed by a complete lack of dilation in response to substance P. Since the P2X receptors desensitize very fast, we constructed a two-log step concentration response curve to αβmetATP, an agonist at P2X receptors. αβmetATP was the most potent agonist (Figure 2(a), pEC50 6.40 ± 0.20, EMAX 117 ± 11%) on the hMMA, while ATPγS (Figure 2(b), pEC50 5.05 ± 0.09, EMAX 173 ± 47%) was less potent than αβmetATP. For the pyrimidine receptors, the most pronounced contraction was observed after activation of the P2Y2 receptor and P2Y4 receptor (Figure 2(c), UTPγS, pEC50 4.73 ± 0.10, EMAX 170 ± 44%). Since UTPγS and ATPγS had similar potency, this indicates that most of the effect is mediated by the P2Y2 receptors. A specific agonist for the P2Y6 receptor caused minor contraction (Figure 2(d), UDPβS, pEC50 5.48 ± 0.41, EMAX 36 ± 14%).
Purinergic-induced contractions of hMMA without endothelium. Concentration-dependent contraction induced by αβmetATP (a), ATPγS (b), UTPγS (c), UDPβS (d) and ADPβS (e) on hMMAs. (f) Arteries were precontracted with 30 mM KCl, and relaxed with CGRP, followed by a concentration response curve to ADPβS. MRS2211, a specific antagonist against the P2Y13 receptor (10 µM), was applied 30 min before performing the concentration dose curve. Data are shown as mean ± SEM, n = 5. (*p < 0.05, **p < 0.01, ***p < 0.001; two-way ANOVA with Bonferroni post-test).
All of the above receptors have previously been investigated in human coronary and cerebral arteries where they induced contraction (22,23), similarly as for the triptans. Therefore, they are not preferable targets for anti-migraine agonists. In contrast, ADPβS, an agonist for the P2Y1 receptor, P2Y12 receptor and P2Y13 receptor, has been shown to be devoid of contractile properties in human coronary and cerebral arteries (22,23). In contrast to the arteries above, in the hMMA, ADPβS caused a minor contraction (Figure 2(e), pEC50 5.99 ± 0.20, EMAX 19 ± 4%). Due to the relatively low EC50 value, corresponding better to the P2Y12 receptor or P2Y13 receptor than the P2Y1 receptor, we applied a P2Y13 receptor antagonist (MRS2211, 10 µM) under two different conditions. First, in a pure contractile experiment, where MRS2211 prevented contraction from baseline (Figure 2(e)). Secondly, we added ADPβS with/without MRS2211 on arteries that were precontracted with 30 mM KCl and had subsequently been relaxed with CGRP (Figure 2)(f). The results show that ADPβS agonism can revert the CGRP-induced relaxation by binding to the P2Y13 receptor, similar to what has been observed with sumatriptan (24).
Effects on in vivo/in situ CGRP release in rat
Intravital microscopy – involvement of the P2Y13 receptor
Our in vitro data show that P2Y13 receptor stimulation induces contraction in the hMMA. To confirm that a P2Y13 receptor agonist can also have effects in vivo, we continued with intravital microscopy in rats, a common method to assess the anti-migraine potential of a compound that is supposed to act on the trigeminovascular system.
Upon intravenous injection of ADPβS (330 µg/kg), we observed a transient decrease in blood pressure (Figure 3(a), 65 ± 3%), which most likely was caused by stimulation of endothelial P2Y1 receptors followed by desensitization (25). Subsequently, a long-lasting increase in blood pressure was observed (Figure 3(a), 23 ± 4%), which, in contrast to the vasodilation, was dependent on the P2Y13 receptor, since the increase was only 7 ± 4% in the presence of MRS2211 (1 mg/kg). MRS2211 did not have any significant effect on the blood pressure per se. The direct effect of ADPβS on the artery was similar to that observed for the blood pressure, with an acute dilation (56 ± 21%) that returned to baseline. In contrast to the blood pressure, we did not observe any direct contraction following the ADPβS (data not shown).
In vivo effects of ADPβS on CGRP-induced dilation of the rMMA. (a) Acute recordings of blood pressure upon infusion of 330 µg/kg ADPβS. The i.v. infusion of ADPβS caused a fast and short-lasting drop in the blood pressure followed by an increase that was prevented by the presence of 1 mg/kg of the specific P2Y1 receptor inhibitor MRS2211. (b) Inhibition of CGRP induced vasodilation of the rMMA by 330 µg/kg ADPβS. (c) In the presence of 1 mg/kg MRS2211, ADPβS does not cause inhibition of periarterial electrically-stimulated CGRP release and rMMA vasodilation. Data are shown as mean ± SEM, n = 6. (*p < 0.05, one-way ANOVA with Dunnett's Multiple Comparison Test). ES: electrical stimulation.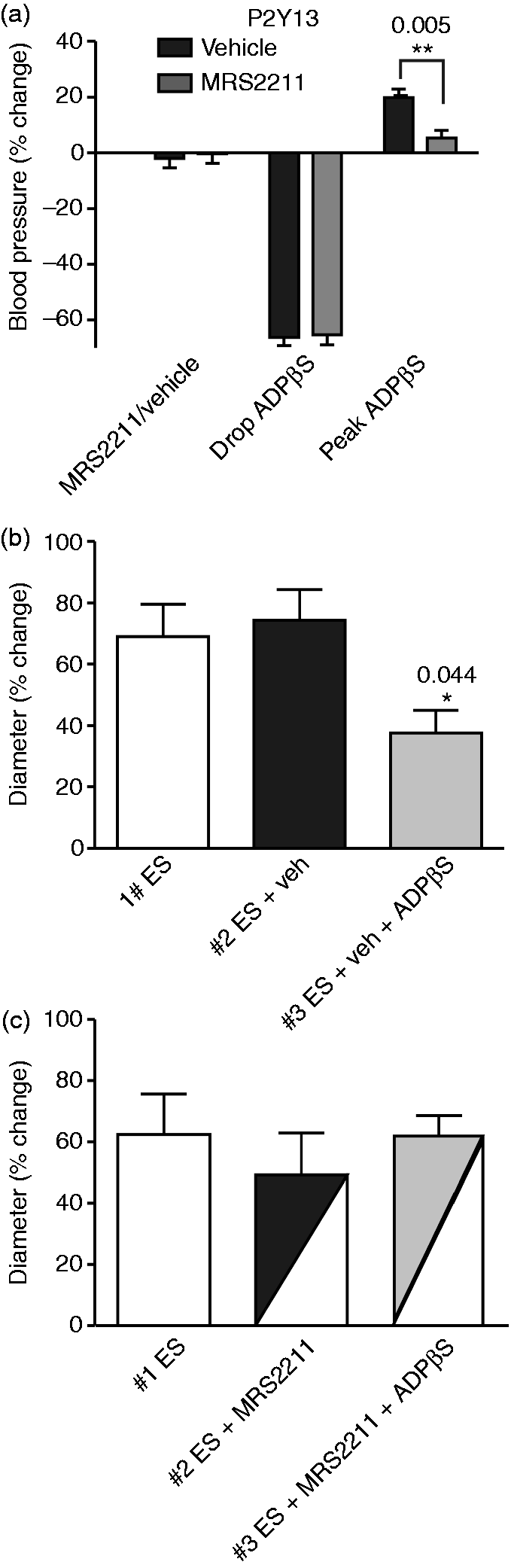
Using periarterial electrical stimulation (ES), which releases CGRP (19,26), the rMMA dilated (Figure 3(b), 69 ± 11%), according to our expectations, and vehicle did not affect this vasodilation (74 ± 10%). In contrast, ADPβS caused a significant inhibition of the dilation (Figure 3(b), 37 ± 7%). Furthermore, MRS2211 (1 mg/kg), which had no significant effects per se (Figure 3(c)), prevented the inhibitory effect of ADPβS and normalized the ES-induced dilation (Figure 3(c), 62 ± 7%). Blood pressure could be of importance in relation to interpreting the diameter increase during electrical stimulation. There were no significant absolute differences during the third ES after ADPβS (130 ± 4 mmHg) compared to ADPβS + MRS2211 (123 ± 5 mmHg).
CGRP release from dura and TG – involvement of the P2Y13 receptor
To dissect the potential site of P2Y13 inhibitory actions, we measured CGRP release ex vivo after individual stimulation of the trigeminal fibres in the dura mater and in the TG isolated from rats. ADPβS significantly inhibited CGRP release from both the dura mater (Figure 4(a) left panels, 172 ± 15 vs. 265 ± 21 pg/ml, p = 0.0003) and from the TG (Figure 4(a) right panels, 162 ± 13 vs. 278 ± 36 pg/ml, p = 0.0141). MRS2211 (10 µM) was without inhibitory effect per se (Figure 4(b)), but abolished the inhibitory response of ADPβS, illustrating that this is a P2Y13-dependent process (Figure 4(c)). Although not within the focus of the current study, ADPβS also inhibited CGRP release from the TNC (Supplementary Figure 1).
In situ effects of ADPβS on CGRP release from the dura or trigeminal ganglion. (a) Addition of ADPβS or vehicle had no effect on CGRP release per se. The addition of 60 mM KCl caused CGRP release from both the dura (n = 5) and the TG (n = 6), which could be inhibited by the presence of 10 µM ADPβS. (b) Incubating with 10 µM of the specific P2Y13 receptor inhibitor MRS2211 had no effects per se (n = 5). (c) 10 µM MRS2211 prevented the inhibition caused by ADPβS (n = 4). Data are shown as mean ± SEM. (*p < 0.05, **p < 0.01, ***p < 0.001; paired Student's t-test).
Intravital microscopy – involvement of the P2X3 receptor
Previous research has shown that P2X3 receptors are co-expressed with CGRP in the trigeminal ganglion (9), and that P2X3 receptor-positive neurons innervate the dura mater (27). Since purinergic research has been mainly focused on the P2X receptors, we continued to investigate the effect of the P2X receptor agonist αβmetATP on the rMMA in vivo, exemplified in a raw trace (Figure 5(a)). αβmetATP (150 µg/kg) caused a strong constriction of the rMMA (Figure 5(b), −70 ± 7%), followed by a recovery dilation (Figure 5(b), 54 ± 10%). This dilation was completely prevented by the addition of the CGRP antagonist olcegepant (Figure 5(b), 1 ± 4%, 100 µg/kg). Furthermore, olcegepant, as expected, prevented dilation both in response to capsaicin (10 µg/kg) and exogenous CGRP (1 µg/kg), but not to substance P (1 µg/kg). For the blood pressure data, we observed a similar response, with an initial increase in blood pressure (Figure 5(c), 34 ± 7%), followed by a reduction of 23 ± 7% (Figure 5(c)). In contrast to the data from the rMMA, the drop in blood pressure was not inhibited by olcegepant. In addition, these data show that the response to exogenous CGRP on the rMMA is unaffected by the presence of ADPβS, unlike the endogenous release (Figure 3(b)).
In vivo effects of αβmetATP on the rMMA. (a) Raw trace from single experiments showing the strong contraction induced by 150 µg/kg of αβmetATP and the following dilation in the presence of vehicle, Olcegepant (100 µg/kg) or ADPβS (330 µg/kg). (b) Inhibition of αβmetATP induced vasodilation of the rMMA by 100 µg/kg olcegepant. (c) Acute recordings of blood pressure upon infusion of 150 µg/kg αβmetATP. The i.v. infusion of αβmetATP caused a fast and short-lasting increase in the blood pressure followed by a short-lasting decrease that was unaffected by the presence of 100 µg/kg olcegepant or 330 µg/kg ADPβS. When the rMMA was not visible, its diameter was set to be 10 µm. Data are shown as mean ± SEM, n = 6. (*p < 0.05, **p < 0.01, repeated measures one-way ANOVA, with Dunnett's Multiple Comparison Test).
Since our data indicate that αβmetATP could trigger CGRP release, we hypothesized that ADPβS, via activation of the P2Y13 receptor, might also be inhibitory on the αβmetATP-induced CGRP release. Surprisingly, we did not observe any significant inhibition on the maximum dilation (Figure 5(b), 35 ± 13% vs. 54 ± 10%). However, there was a significant delay in the time to peak dilation, which is also visible in the illustrative raw-trace (Figure 5(a), 216 ± 27 sec vs. 147 ± 8 sec, p = 0.0307), illustrating the potential involvement of the P2Y13 receptor in αβmetATP-induced CGRP release.
CGRP release from dura mater and TG – involvement of the P2X3 receptor
The dilation observed after constriction of the rMMA could be caused by: i) CGRP release triggered at the synapse or ii) antidromic CGRP release triggered by P2X receptors in the TG. We therefore investigated the effect of αβmetATP on CGRP release in the dura mater and the TG. We observed that αβmetATP did not cause any CGRP release from the dura mater above baseline values (Figure 6 left panel, 31 ± 5 vs. 32 ± 5 pg/ml), in contrast to when it was applied to the TG (Figure 6 right panel, 58 ± 7 vs. 102 ± 15 pg/ml, p = 0.0073).
In situ effects of αβmetATP on CGRP release from the dura or trigeminal ganglion. The addition of 5 µM αβmetATP caused a strong release of CGRP from the trigeminal ganglion but not from the dura. 60 mM KCl caused CGRP release from both the dura and the TG, which was reduced following the release induced by αβmetATP. Data are shown as mean ± SEM, n = 5. (*p < 0.05, **p < 0.01; paired Student's t-test).
Immunohistochemistry on hTG and hMMA: Localization of P2Y13 receptor and P2X3 receptor, co-localization with CGRP
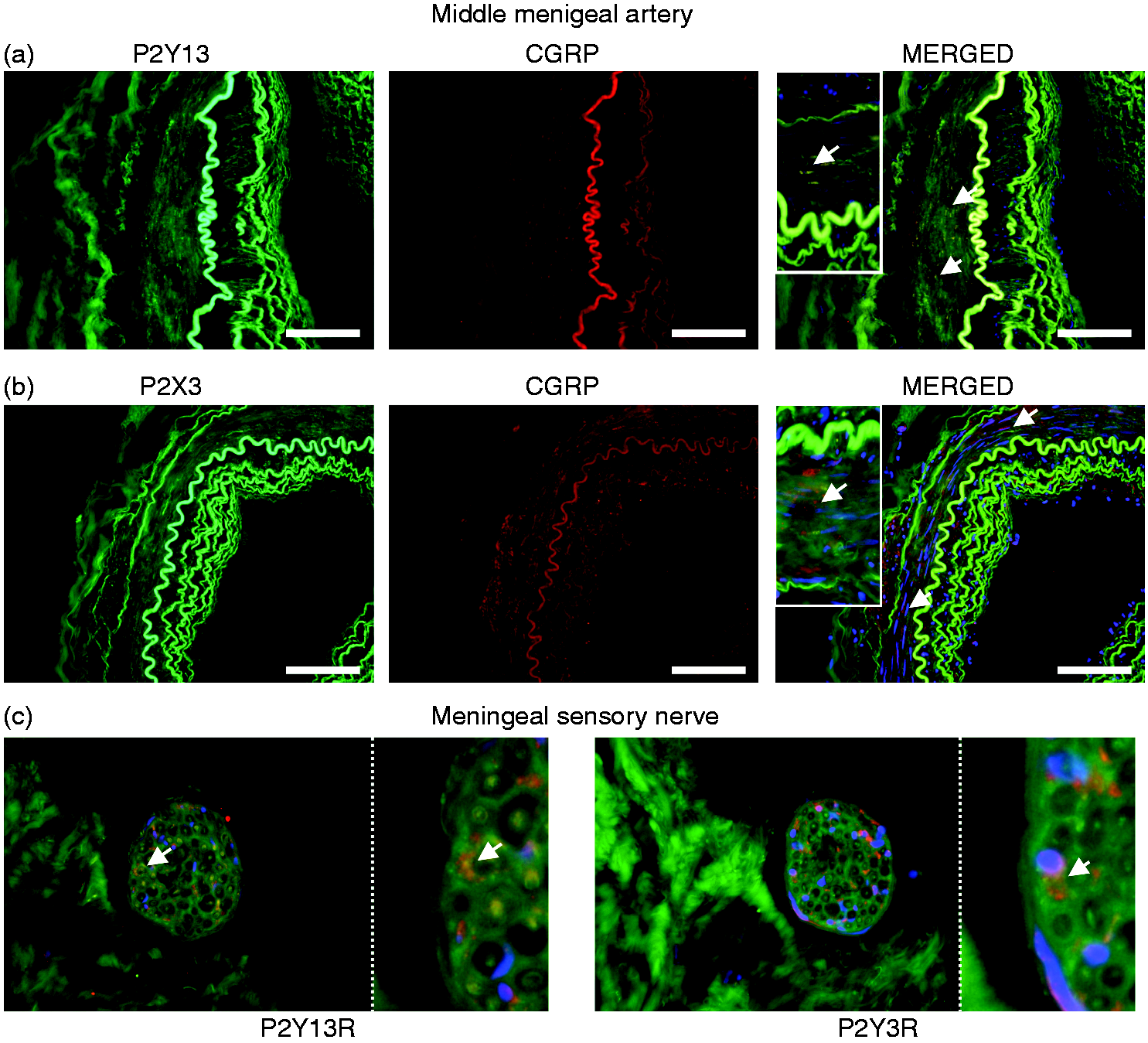
We have here presented strong functional evidence for the involvement of the P2Y13 receptor and P2X receptor in in vivo and in situ rat models. Similar experiments are not possible in humans, but we had access to human tissue. Although the samples were obtained post mortem and could have been subject to potential changes in protein expression, we investigated whether similar receptor components are present in the human trigeminovascular system. Although sparsely expressed, we did detect neurons expressing both the P2Y13 receptor and CGRP in the human trigeminal ganglion (Figure 7(a)). In contrast, the P2X3 receptor is strongly expressed together with CGRP (Figure 7(b)). In the hMMA, CGRP and the P2Y13 receptor co-localized, in what appear to be C-fibre nerve endings (Figure 8(a), white arrows), whereas the P2X3 receptor is sparsely, if at all, present in the hMMA (Figure 8(b)). In one of the samples of hMMA, we observed a meningeal nerve (Figure 8(c)). Here, it is even more evident that the P2Y13 receptor co-localizes with CGRP in the C-fibres, but this is not the case for the P2X3 receptor (white arrows).
Immunohistochemical localization of P2Y13 receptor and P2X3 receptor in the human trigeminal ganglion. (a) Representative expression of P2Y13 receptor (green) and CGRP (red) in the human trigeminal ganglion co-localizes in some cells (merged). (b) Representative expression of P2X3 receptor (green) and CGRP (red) in the human trigeminal ganglion co-localizes in several of the cells (merged). DAPI was used to stain the nuclei for the merged picture. Scalebar is 100 µM. Immunohistochemical localization of P2Y13 receptor and P2X3 receptor in the hMMA. (a) Representative expression of P2Y13 receptor (green) and CGRP (red) in the hMMA, co-localizes in nerve endings, see arrows (merged). (b) Representative expression of P2X3 receptor (green) and CGRP (red) in the hMMA did not co-localize at the nerve endings, see arrows (merged). (c) Insert of a nerve following the hMMA further illustrates the co-localization of P2Y13 receptor (green) and CGRP (red), but not any co-localization of CGRP with the P2X3 receptor. DAPI was used to stain the nuclei for the merged picture. Scalebar is 100 µM.
Discussion
Direct effects on the MMA
Until CGRP receptor antagonists were developed, all acutely acting specific anti-migraine drugs induced direct vasoconstriction. Although direct vasoconstriction of the MMA may not be an essential requirement in treating migraine (1), it still might play a relevant role (28). In addition, the MMA may serve as a proxy of the trigeminal system, where potential anti-migraine compounds can be tested. In the current study, we started with a characterization of purinergic receptors on the hMMA. The data show that purinergic receptors have important effects. Particularly, their activation on the endothelium may induce strong dilation (Figure 1), while their activation in smooth muscle cells may induce a strong contraction (Figure 2). Only the contractile responses can be compared to our previously obtained data in the rat, as there were no endothelial responses in the rMMA due to their limited diameter (29).
Vasoactive properties of purinergic ligands on the hMMA
The P2X receptor agonist αβmetATP caused the most potent contraction in the hMMA (pEC50 6.40 ± 0.20, Figure 2(a)). This is expected, as the P2X1 receptor is known to mediate potent contractions in smooth muscle cells (30). The main pyrimidine receptor in the hMMA is the P2Y2 receptor (Figure 1(b) and Figure 2(b)/(c)). When activated in the endothelium of the hMMA, it causes a strong vasodilation, with higher potency than in the human internal mammary artery (31). The P2Y2 receptor is also the receptor that leads to the strongest contraction of the MMA, similar to human coronary and omental arteries (22,23). Activation of the P2Y6 receptor caused a pure contractile effect (Figure 1(c) and Figure 2(d)). The EMAX was not very high, which is similar to the human omental artery (22). In human coronary arteries, there was no contraction to UDPβS (23), which contrasts to the cerebral arteries, where UDPβS is a strong constrictor (22). ADPβS was a strong vasodilator (Figure 1(a)), which is typically attributed to the P2Y1 receptor (31). The contraction to ADPβS in the hMMA was potent, but with a relatively low EMAX (Figure 2(e)). In conclusion, the functional purinergic profile of the hMMA is most similar to human omental arteries and shares strong similarities to the coronary arteries. The profile in hMMA contrasts with that in human cerebral arteries, where the main difference is strong contractility to UDPβS (P2Y6 receptor agonist). Contractions to ADPβS are not described in any of the other human arteries, making the hMMA unique.
Focus on the P2Y13 receptor in the hMMA
We were further particularly interested in ADP and its stable analogue ADPβS, since it is known that this does not cause contraction in the human coronary or cerebral vasculature (22,23). The side effect of coronary vasoconstriction is a current caveat of the current acutely acting antimigraine treatments such as the triptans (32). The contractile response to ADPβS in hMMA is small but significant. Furthermore, the response to ADPβS on the hMMA seems to be mediated purely by the P2Y13 receptor, as it is blocked by its antagonist MRS2211 (Figure 2(e)/(f)), at a concentration that should be devoid of effects on the P2Y1 and P2Y12 receptors (33). Although the contraction to ADPβS was minor, ADPβS and P2Y13 receptor activation could reverse the dilation caused by CGRP (Figure 2)(f). This has a strong link to anti-migraine potential, and the data are very similar to what has been observed for the triptans (24).
Comparison between hMMA and rMMA
In our previous study on rMMA, ADPβS caused a strong contraction compared to the current data on the hMMA. We did not explore the functional pharmacological identity of the ADPβS response in the rat study, but we detected a strong P2Y13 receptor signal in the rMMA by PCR, combined with some P2Y1 receptor expression, but no expression of the P2Y12 receptor (29). Regarding the difference in contraction between rMMAs and hMMAs, it appears that in both human and rat, fast acting and short-lasting effects are mediated via ATP on P2X receptors. In humans, long-term effects of purines are effectuated by ATP on the P2Y2 receptor, in contrast to rats where ADP is acting on the P2Y1 receptor (29). For pyrimidines, UTP through P2Y2 receptor activation is important in humans, while the breakdown product UDP acts on the P2Y6 receptor in rodents. It therefore seems that the human artery is more sensitive to ATP. In conclusion, the purinergic contractions in hMMA are caused by ATP/UTP, in contrast to the rMMA, which contracts to their breakdown-products UDP/ADP. Nevertheless, in vivo, the response is most likely similar, as the breakdown of UTP/ATP into UDP/ADP is nearly instantaneous (34), exemplified by studies on P2Y2 receptor and P2Y6 receptor knock-out mice (35) and functional data from rat meningeal arteries (36).
Trigeminovascular effects
In the light of the recent advances in migraine research, investigating the in vivo effects of potential antimigraine compounds on CGRP release is a highly relevant approach. We therefore used an established method to study the in vivo effects of ADPβS and αβmetATP, which activate P2Y13 and P2X receptors, respectively.
Inhibitory effects of P2Y13 receptor agonism
Our data show that ADPβS, through activation of the P2Y13 receptor, inhibits vasodilation caused by periarterial electrical stimulation (Figure 3(b)/(c)). Periarterial stimulation has been shown to cause in vivo release of CGRP (19,26). Therefore, our experiments are the first to show that P2Y receptors, coupled to Gi proteins, can have prejunctional inhibitory effects on CGRP release in vivo. Similar observations have been made on other prejunctional nerve endings, where P2Y13 receptor activation was demonstrated to inhibit noradrenaline release from sympathetic neurons (16) and cholinergic transmission on the neuromuscular junction (17). Importantly, we also show the direct inhibitory effect of ADPβS on CGRP release from both the dura and the TG in situ (Figure 4(a)). ADPβS is activating the P2Y13 receptor, as the specific P2Y13 receptor inhibitor MRS2211 was without effects per se but reversed the inhibitory effect of ADPβS (Figure 4(b)/(c)). This is similar to the experiments by Honey et al., where an adonsine A1 receptor antagonist (DPCPX) blocked the inhibitory effect of an A1 agonist (GR79236) on meningeal vasodilation but had no effects per se (37). Since inhibiting CGRP release (or CGRP signalling) is now believed to be the most important hallmark of all acute anti-migraine treatments (1), our data clearly demonstrate the therapeutic potential of a specific P2Y13 receptor agonist.
Trigeminal activation by P2X receptor activation
αβmetATP injections caused an activation of the trigeminovascular system, starting with a strong, short-lasting contraction of the rMMA, followed by a relaxation (Figure 5(a)). The relaxation is mediated by CGRP, as demonstrated by the blockade induced by the CGRP receptor antagonist olcegepant (Figure 5(b)). This is highly similar to the experiment by McCulloch and colleagues that was performed over 30 years ago, who showed that trigeminal nerve lesion prevented the relaxation following a constriction of the meningeal artery (38). Our data show that αβmetATP-induced activation of P2X receptors on the TG is the most likely mechanism of action, leading to CGRP release (Figure 6). Indeed, Yegutkin et al. showed that αβmetATP triggered trigeminal nerve fibre activity (36) and Eroli et al. have shown that αβmetATP evokes neuronal firing in trigeminal cultures (39). Therefore, stimulation of P2X3 receptors using αβmetATP could be a model of trigeminal activation, particularly for antidromic CGRP release occurring at the MMA. This is supported by: i) The relatively long time from stimulation until observable dilation (Figure 5(a)), as the signal to release CGRP might travel from the TG and ii) the ability of αβmetATP to trigger CGRP release only from the TG and not in the dura mater (Figure 6).
Expression in human TG and clinical relevance
We investigated whether the purinergic receptors found to be important in the rat trigeminovascular system are also expressed in the human TG. The P2X3 receptor was strongly expressed in CGRP-ergic neurons (Figure 7(a)), similar to what has been shown before in rats (9). There are also neurons that are positive for both the P2Y13 receptor and CGRP, although we only sporadically observed these (Figure 7(b)). In addition, we investigated the hMMA and its innervation. Here we found that, in contrast to the TG, the P2Y13 receptor is co-localized with CGRP (Figure 8(a)), in contrast to the P2X3 receptor (Figure 8(b)). Combined with the findings in the TG, these data match the functional data that we observed in the rat. Both the functional data and the immunohistochemistry indicate that P2X3 receptor activation and expression mainly occurs in the TG (Figures 5/6 and 7/8), and that P2Y13 receptor inhibition and expression can both exist in the TG (Figures 3/4 and 7) and on the nerve endings at the dura/MMA (Figures 3/4 and 8). Combined, the above findings suggest that there are similarities in the purinergic aspects of the neurovascular system between humans and rats.
The clinical applications and potential anti-migraine effects need to be further investigated; for this purpose, there is a particular need for the development of specific agonists. The main concern regarding the use of agonists based on ADP is their potential thrombic activity. ADP activating P2Y1 or P2Y12 receptors is considered a relatively weak platelet aggregation agonist by itself (we did not observe any acute thrombic events in the current experiments). Unfortunately, ADP still works as an amplifier of platelet aggregators such as thrombin (40), which is not compatible with treatment of patients. This potential side-effect should not be an issue for a specific P2Y13 receptor agonist.
Conclusion
In the current study, we performed a thorough investigation of the purinergic receptors in the hMMA and compared it to our previous data on the rMMA. Interestingly, the expression and regulation of contractile responses are mediated by different receptors in human and rat. P2Y2 receptors (activated by ATP/UTP) are mediating most of the G-protein coupled purinergic function in hMMA compared to a combined effect of the P2Y1 receptor (ADP) and P2Y6 receptor (UDP) in rMMA.
The P2Y13 receptor was important in counteracting the effects of CGRP on the hMMA and P2Y13 receptor activation also inhibited CGRP release, both in vivo and in situ. Therefore, the P2Y13 receptor could be a novel anti-migraine target, particularly when considering the intracellular pathway activated by this receptor in light of the migraine pathology (14). Furthermore, activation of the P2X3 receptor caused specific CGRP release originating from the TG and a consequent olcegepant-sensitive dilation at the rMMA. Our findings contribute to further understanding of the pathophysiology of migraine. To investigate the full clinical potential of targeting the P2Y13 receptor, a pharmacologically specific agonist is clearly needed.
Supplemental Material
Supplemental material for Exploration of purinergic receptors as potential anti-migraine targets using established pre-clinical migraine models
Supplemental Material for Exploration of purinergic receptors as potential anti-migraine targets using established pre-clinical migraine models by Kristian A Haanes, Alejandro Labastida-Ramírez, Frank W Blixt, Eloisa Rubio-Beltrán, Clemens M Dirven, Alexander HJ Danser, Lars Edvinsson and Antoinette MaassenVanDenBrink in Cephalalgia
Footnotes
Key findings
P2Y13 receptor activation can counteract the effects of CGRP on the hMMA.
ADPβS activating the P2Y13 receptor inhibited CGRP release, both in vivo and in situ, and could therefore be a novel anti-migraine target.
Activation of the P2X receptor caused specific CGRP release originating from the TG and a consequent olcegepant-sensitive dilation at the rMMA.
Acknowledgements
We would like to thank Professor János Tajti (University of Szeged, Hungary) for providing us with human trigeminal tissue. We would further like to thank Professor Ivana Novak (University of Copenhagen) for constructive criticism on the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: KAH was supported by a fellowship from the International Headache Society. AMVDB was supported by the Netherlands Organization for Scientific Research (Vidi grant 917.113.349), ALR and ERB were supported by Consejo Nacional de Ciencia y Tecnología (CONACyT; fellowships No. 410778 to ALR and No. 409865 to ERB; Mexico City). Funders had no role in study design.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.