
Abstract
Background
Safety findings from a Phase 2b study of galcanezumab, a humanized monoclonal antibody against calcitonin gene-related peptide, for prevention of migraine (NCT02163993) are reported here.
Methods
Patients aged 18–65 years with episodic migraine were evaluated in this multicenter, double-blind, randomized study. After randomization, 410 patients were administered 5, 50, 120 or 300 mg of galcanezumab or placebo subcutaneously once every 4 weeks for 12 weeks, followed by a post-treatment off-drug period lasting 12 weeks.
Results
Treatment-emergent adverse events (TEAEs) were primarily rated as mild to moderate. Serious adverse events reported in galcanezumab dose groups were appendicitis, Crohn’s disease, suicidal ideation, and congenital ankyloglossia in an infant of a paternal pregnancy; each of these were reported by one patient. Adverse events leading to discontinuation with galcanezumab treatment were abdominal pain, visual impairment, and upper limb fracture, each reported by one patient. Treatment-emergent injection-site reactions were reported significantly more frequently (p = 0.013) with galcanezumab (13.9%) than with placebo (5.8%). Injection-site pain was the most common injection-site reaction (galcanezumab 11.4%; placebo 2.9%, p = 0.004). Upper respiratory tract infection (galcanezumab 10.0%; placebo 8.8%) and nasopharyngitis (galcanezumab 7.0%; placebo 2.2%) also occurred more frequently with galcanezumab treatment. Potential hypersensitivity events were reported at similar frequencies in galcanezumab (3.3%) and placebo (5.1%) groups. Incidence of treatment-emergent anti-drug antibodies in galcanezumab dose groups (4.6% of patients during treatment period) did not appear to have any meaningful effects on safety, the pharmacokinetics of galcanezumab, or its ability to bind to the target ligand.
Conclusion
The results from this 3-month Phase 2b study support the initiation of larger Phase 3 trials of longer duration.
Introduction
Galcanezumab (LY2951742) is a humanized monoclonal antibody that potently and selectively binds to calcitonin gene-related peptide (CGRP) and is being developed for the prevention of migraine. CGRP is a 37-amino acid neuropeptide that is found throughout the trigeminal system and in the central nervous system (1,2). CGRP has been implicated in the pathogenesis of migraine through its ability to induce the release of local inflammatory mediators and directly transmit nociceptive information from intracranial blood vessels to the nervous system (3,4). CGRP is also a potent vasodilator (5).
Migraine is a debilitating neurological disease with a significant socioeconomic burden and an estimated worldwide prevalence of 14.7% (7,8). Although there are multiple treatment options for migraine prevention, not all patients respond to these existing therapies (9,10). Thus, new treatment options are needed, and galcanezumab belongs to a novel class currently in development (11,12). It is important to establish the safety profile of this new class before it enters widespread use for long periods of time and in a population that is relatively healthy.
Galcanezumab was previously studied in a proof-of-concept Phase 2a study in 218 patients with migraine where it was administered at a dose of 150 mg every 2 weeks for 12 weeks (13). In that study, none of the 107 patients treated with galcanezumab discontinued due to an adverse event (AE). Treatment-emergent injection-site pain or erythema, upper respiratory tract infections, and abdominal pain were the treatment-emergent adverse events (TEAEs) reported more frequently with galcanezumab treatment compared with placebo. None of the patients showed clinically important changes in laboratory parameters, electrocardiograms (ECGs), or vital signs (13).
A Phase 2b, randomized, placebo-controlled study was conducted to evaluate the safety and efficacy of galcanezumab 5, 50, 120, and 300 mg, administered every 4 weeks (Q4W) in patients with migraine (14). Here we provide a detailed description of the safety findings from this study.
Methods
Study design and patients
This was a Phase 2b, multicenter, double-blind, randomized, placebo-controlled study that evaluated the efficacy and safety of four galcanezumab doses in preventing migraine headache. There were four study periods (SPs): Screening and washout period (SPI); prospective 4-week baseline period (SPII); 12-week treatment period (SPIII); and 12-week post-treatment period, which was a safety follow-up period (SPIV) during which patients did not receive the study drug or placebo (Figure 1). Patients were randomized (2:1:1:1:1) to receive placebo or one of the four galcanezumab doses (5, 50, 120, or 300 mg). During the treatment period, each dose of galcanezumab or placebo was administered as two 1.5 mL subcutaneous injections, Q4W at weeks 0, 4, and 8 for a total of three dose administrations. During the post-treatment period, patients no longer received injections of the study drug; any references to the treatment group when presenting results for post-treatment period reflect patients’ previous treatment assignment. When greater clarity regarding dose group is required, terminologies such as galcanezumab_all or galcanezumab-treated (all doses combined) or galcanezumab_5 mg (individual dose group) are used.
Study design of placebo-controlled dose-ranging Phase 2b study of galcanezumab in patients with episodic migraine (NCT02163993). aElectronic patient-reported outcomes using IVRS reporting were completed daily during baseline, treatment and post-treatment periods. bBetween office visits, sites were required to have a telephone visit at visits 4, 6, 10, and 12 to assess spontaneously reported AEs. cVisit 8 was to obtain PK and CGRP blood samples and assess safety. Abbreviations: AEs: adverse events; CGRP: calcitonin gene-related peptide; IVRS: interactive voice response system; PK: pharmacokinetics; SP: study period.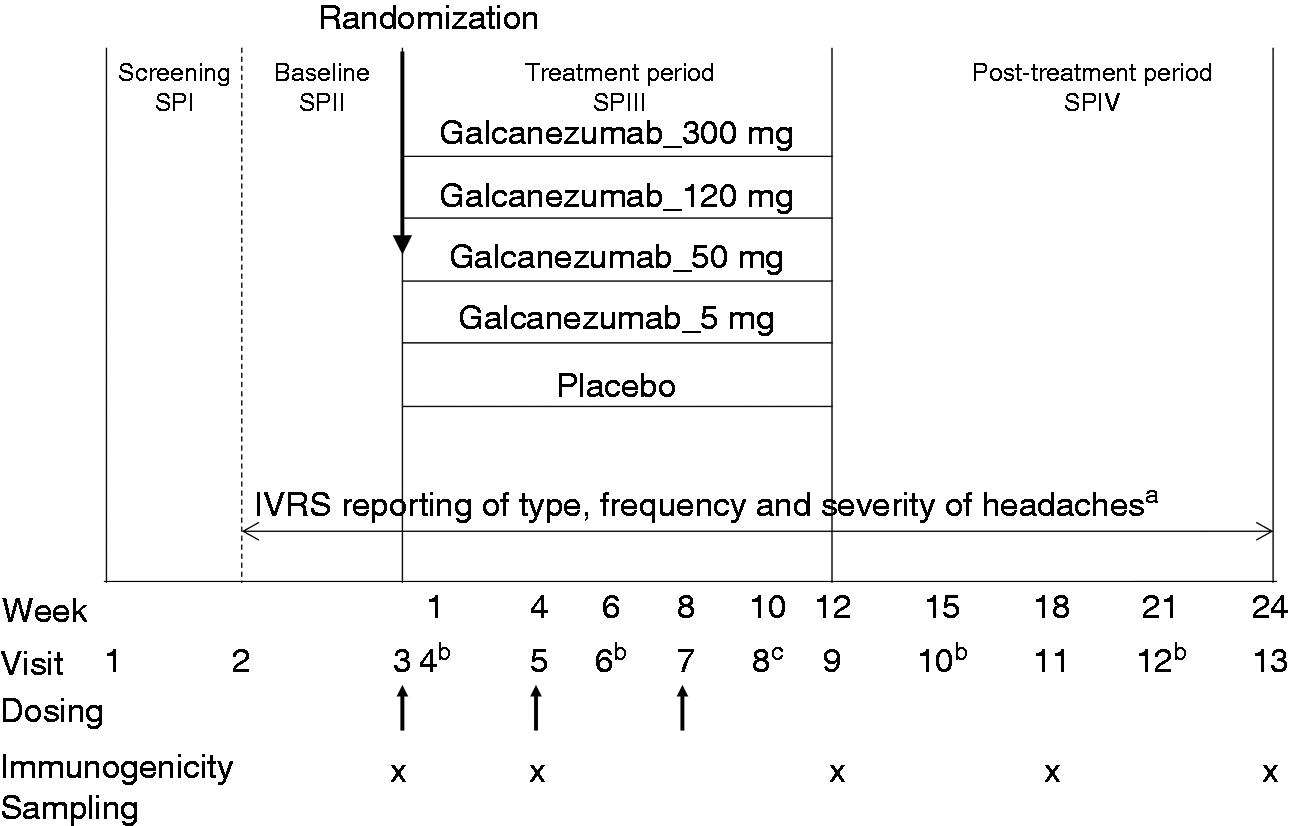
Study participants consisted of male and female patients aged 18–65 years with a history of episodic migraine as described in the International Headache Society International Classification of Headache Disorders-3 beta guidelines (15) and experiencing 4–14 migraine headache days in a 28-day period during SPII. The key safety-related exclusion criteria were significant active psychiatric disease (unless stable and expected to remain stable), active suicidal ideation or behavior in the one month prior to enrollment, recent history of serious cardiovascular events, abnormalities in ECGs compatible with acute cardiovascular events and/or serious cardiovascular risk, body mass index (BMI) ≥40 kg/m2 (because of potential for cardiovascular risk), and clinically significant laboratory abnormalities.
The study was approved by the respective institutional review boards for each of the study sites and written consent was obtained from all patients in the study prior to participation.
Prior and concomitant therapy
Use of medications or procedures for preventive treatment of migraine was not allowed for at least 30 days prior to Visit 2 (baseline visit) and until the end of the treatment period. Patients could take acute treatments such as triptans, ergotamines, acetaminophen, aspirin, and non-steroidal anti-inflammatory drugs, but not opioids or barbituates.
Evaluation of adverse events
Safety assessments at each office visit included spontaneously reported AEs, blood pressure, pulse, the Columbia Suicide Severity Rating Scale (C-SSRS), and concomitant medications. TEAEs were events that first occurred or worsened during the treatment phase. Suicidality was not an AE of interest but was assessed as part of the standard evaluation of a neurological drug. AEs during post-treatment/safety follow-up period refers to AEs that first occurred or worsened during the post-treatment period. Based on Phase 2a data (13), AEs of interest included potential hypersensitivity events, injection-site AEs, and upper respiratory tract infections (URTIs). Injection-site AEs and upper respiratory tract infections (URTI) were identified using all preferred terms from the high-level terms of “injection-site reaction” and “upper respiratory tract infections” from the Medical Dictionary for Regulatory Activities (MedDRA®) version 17.1. Potential hypersensitivity events were identified using the broad and narrow standardized MedRA queries (SMQs) of anaphylactic reactions, angioedema, severe cutaneous adverse reaction, and hypersensitivity.
Vital signs, weight, and electrocardiograms
Blood pressure and pulse measurements were taken in triplicate at approximately 30–60 second intervals; weight and temperature were also assessed. A single 12-lead ECG was collected with patients in the supine position, at screening, week 10, and week 24 or early termination. ECGs were evaluated by the primary investigator and over-read by a central cardiologist.
Clinical laboratory evaluations
Laboratory assessments were completed at baseline, week 12, and week 24 or early termination visit. The number of patients with abnormal laboratory values at any time was assessed. Covance reference ranges were used for determining an abnormal result (Covance Central Laboratory Services, Indianapolis, IN, USA).
Immunogenicity analyses
Samples for immunogenicity testing were collected at weeks 0, 4, 12, 18, and 24 or at early termination visit; week 24 was part of a post-hoc analysis that comprised samples from only galcanezumab-treated patients.
Anti-drug antibodies (ADA) and accompanying titers were assessed using a validated assay (Online Supplementary Information S1). Treatment-emergent ADA positive (TEADA+) status was defined as follows:
A negative baseline ADA result and a subsequent positive post-baseline ADA result with a titer ≥20 A positive baseline ADA result and a subsequent positive post-baseline ADA result with a ≥ 4 fold increase in titers
Treatment-emergent ADA negative (TEADA−) status was defined as ADA undetected throughout the study, or ADA detected but not considered treatment-emergent. ADA positive samples were further evaluated in a competitive ligand-binding neutralizing assay for detecting neutralizing antibodies. Patients with status of TEADA+ and TEADA− and who discontinued due to an AE, experienced a serious adverse event (SAE), a potential hypersensitivity event, and/or an injection-site reaction were also summarized.
Pharmacokinetic and pharmacodynamic sampling and analyses
Venous blood samples were collected from patients to determine the serum concentrations of galcanezumab and plasma concentrations of CGRP using validated bioanalytical methods (Online Supplementary Information S1). The galcanezumab and CGRP concentrations collected at weeks 4, 12, 18 and 24 were stratified by dose regimen and assessed in patients classified as TEADA+ and TEADA− during the duration of the trial.
Statistical analyses
All safety analyses were based on the intent-to-treat (ITT) population; that is, randomized patients who received at least one injection of the study drug. For AEs, vital signs, laboratory assessments and ECGs, treatment-emergent events or values during the treatment period were analyzed using all visits prior to first injection (visits 1–3) as baseline; events or values during the safety follow-up period were analyzed using all visits prior to this period (visits 1–9 or weeks 1–12) as baseline. For repeated continuous measures including change in vital signs from baseline (last non-missing value of visits 1–3), a restricted maximum likelihood-based mixed effects repeated measures (MMRM) analysis was used for (a) the treatment period only and (b) treatment and post-treatment periods combined. The model included treatment, pooled investigative site, visit, treatment-by-visit interaction, baseline, and baseline-by-visit interaction. For other continuous measures (including laboratory measures and ECGs), changes from the last baseline value to last-observation-carried-forward (LOCF) endpoint during the double-blind treatment phase were assessed using an analysis of covariance (ANCOVA) model with terms for treatment, baseline, and pooled investigator. Comparisons between treatment groups for all categorical safety measures used Fisher’s exact test. This study is not powered to detect statistical significance for differences between groups for rare adverse events. Therefore, results from this study need to be understood along with clinical meaningfulness of each descriptive measure.
Results
Patient disposition and demographics
Patient disposition and demographics for this study have been described in detail elsewhere (14). Briefly, of the 410 patients who were randomized and received the study drug, 273 were assigned to the galcanezumab dose groups and 137 to the placebo group. Overall, 238 galcanezumab-treated patients (87%) and 120 placebo-treated patients (88%) completed both treatment and post-treatment periods. Patients were primarily female (83%) and white (75%), with a mean age of 40 years. The most common concomitant medications (used by ≥5% of all patients) in either the treatment or post-treatment period were nonsteroidal anti-inflammatory agents, vitamins, acetaminophen, acetylsalicylic acid, triptans, and allergy medicines.
Serious adverse events
Summary of adverse events during treatment and post-treatment periods. a

No deaths were reported in the study. One SAE, ankyloglossia congenital reported in the GMB_300 mg group after database lock. bPatients did not receive the study drug during the post-treatment period. cBaseline includes visits 1–9. Abbreviations: AE: adverse event; DCAE: discontinued due to an adverse event; GMB: galcanezumab; N: number of patients who received at least one injection (for treatment period) or number of patients who entered post-treatment period; n: number of patients within each specific category of treatment-emergent adverse events; TEAE: treatment-emergent adverse events; SAE: serious adverse events.
None of the SAEs were considered related to galcanezumab by the investigator or by the study sponsor.
Adverse events leading to discontinuation
Discontinuation due to an AE was reported by three galcanezumab-treated patients and none with placebo (Table 1). During the treatment period, one patient (galcanezumab_5 mg) discontinued due to visual impairment, reported as “right eye spots”. The investigator judged the AE to be related to treatment; however, a consulting ophthalmologist attributed the AE to migraine. Another patient (galcanezumab_300 mg) discontinued due to worsening of preexisting abdominal pain. The patient subsequently reported a prolapsed uterus and urinary tract infection during the post-treatment period. This AE was not considered related to the study drug by the investigator. During the post-treatment period, a patient (galcanezumab_50 mg) discontinued due to an AE of upper limb fracture, which was also considered unrelated to the study drug.
Adverse events
Treatment-emergent adverse events during treatment period and adverse events during the safety follow-up or post-treatment period (baseline period: Visits 1–9).
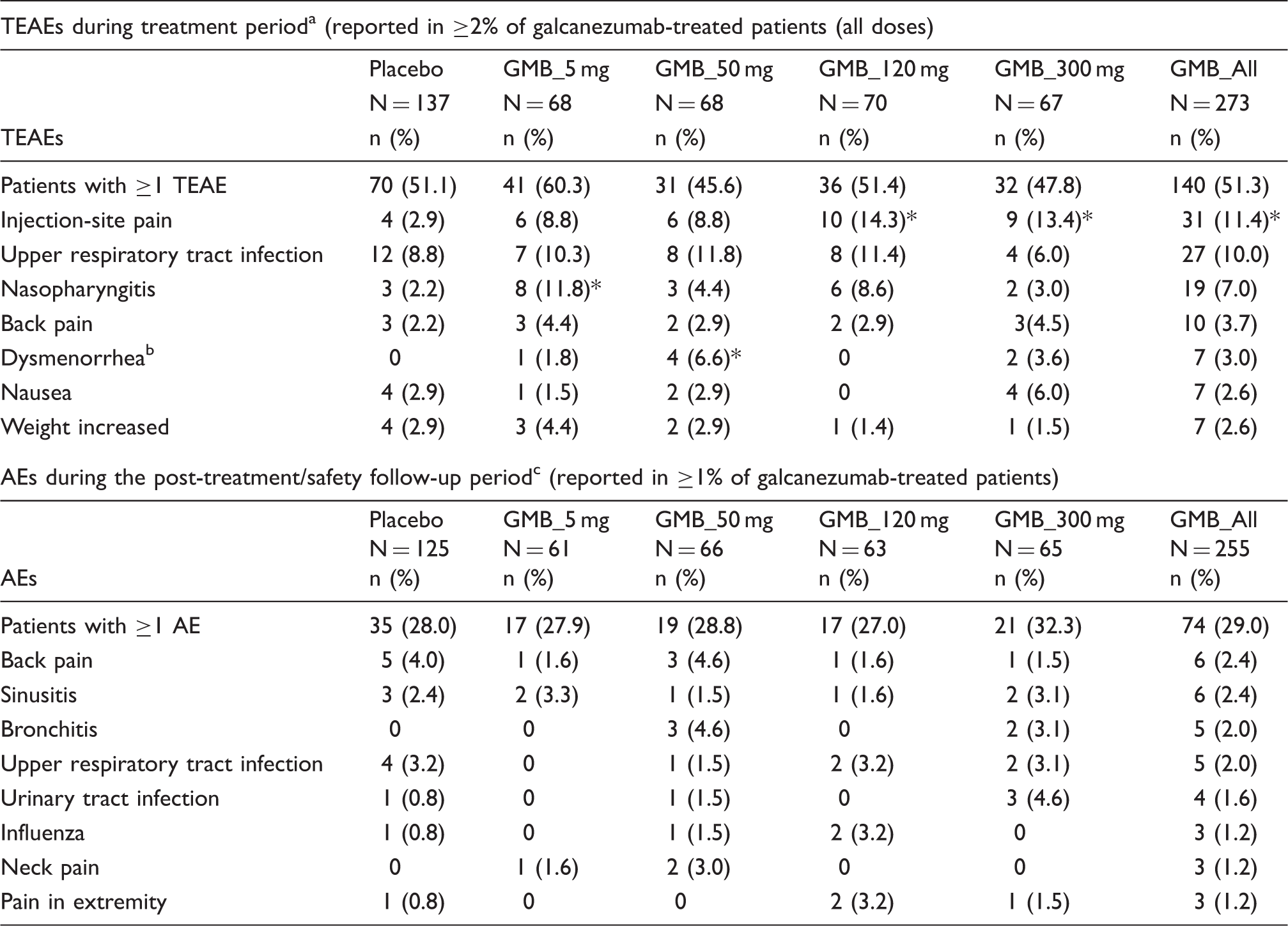
During treatment period, dysgeusia was reported at statistically significantly higher frequency (n = 3, 4.4%) in the galcanezumab_5 mg group compared with placebo (0%) (p = 0.04); none of the other galcanezumab dose groups reported dysgeusia; bfor females only: N = 109 for placebo; N = 55 for GMB_5 mg; N = 61 for GMB_50 mg; N = 59 for GMB_120 mg; N = 56 for GMB_300 mg. cPatients did not receive the study drug during the safety follow-up or post-treatment period; *p-value ≤ 0.05 compared with placebo. Abbreviations: AE: adverse event; GMB: galcanezumab; mg: milligram; N: number of intent to treat patients; n: number of patients with treatment-emergent adverse events; TEAE: treatment-emergent adverse event.
Injection-site adverse events
Incidence of injection-site treatment-emergent adverse events during treatment period reported in at least one galcanezumab-treated patient (any dose group).

*Injection-site erythema, hematoma, induration, pruritus, reaction, and urticaria were each reported by one patient in the galcanezumab_all groupp-value ≤ 0.05 compared with placebo. Abbreviations: GMB: galcanezumab; mg: milligram; N: number of intent to treat patients; n: number of patients with treatment-emergent adverse events; TEAE: treatment-emergent adverse event.
Upper respiratory tract infections
During the treatment period, nasopharyngitis was reported significantly more frequently for galcanezumab_5 mg (11.8%) compared to placebo (2.2%), whereas significantly more patients on placebo reported sinusitis (3.7%) than patients on galcanezumab (galcanezumab_all: 0.7%). No dose correlation was observed for any of the URTI events (Online Supplementary Information S2). During the post-treatment period, the incidence of URTI events was low (<4% in any galcanezumab dose group), and the type and frequency of events did not differ based on prior treatment assignment. During both treatment and post-treatment periods, the URTI events were reported as mild or moderate. During the treatment period, nasopharyngitis reported by one patient (galcanezumab_300 mg) was the only URTI event noted as not resolved in the galcanezumab_all group; all URTI events resolved during the post-treatment period.
Potential hypersensitivity events
During the treatment period, potential hypersensitivity events occurred in 3.3% of galcanezumab-treated patients and 5.1% of placebo-treated patients (Online Supplementary Information S2). None of the events were reported immediately after injection (<60 min) of study drug injection, and none were reported as severe or in association with symptoms suggestive of anaphylaxis. The incidence of potential hypersensitivity events during the post-treatment period was approximately 1% and did not differ based on treatment received during the previous period.
Columbia-Suicide Severity Rating Scale
Suicidal ideation was reported in one patient (Table 1) and is described in the SAE section; none of the other patients in the study reported suicidal ideation. No suicidal behaviors were reported in the study.
Clinical laboratory evaluations
During both treatment and post-treatment periods, mean changes in laboratory evaluations were generally minimal, and mean values remained well within normal range. Significantly more (p = 0.045) patients in the galcanezumab_50 mg group had treatment-emergent abnormal urine ketones (6.4%) compared to placebo (0.8%), although this was not observed with the other doses or in the pooled galcanezumab group, and did not appear to be clinically meaningful. There were no other statistically significant differences or any clinically important differences between galcanezumab dose groups and placebo. Among patients with normal hepatic laboratory values at baseline, no patient showed abnormal hepatic laboratory values during the treatment or post-treatment period.
Immunogenicity
Number and percentage of patients with anti-drug antibodies and treatment-emergent anti-drug antibodies.

Baseline results are prior to administration of any study treatment. bIncludes 18 patients who became TEADA+ at week 18. cWeek 24 analyses were post hoc and no placebo samples were analyzed. dFor GMB_300 mg and GMB_All, N = 66 and N = 265, respectively. eIncludes 51 patients who became TEADA+ at week 24. Abbreviations: ADA: anti-drug antibodies; GMB: galcanezumab; mg: milligram; N: total number of patients in the treatment arm; n: number of patients with treatment-emergent anti-drug antibodies; TEADA: treatment-emergent anti-drug antibodies.
From week 4 to week 18, the proportion of galcanezumab-treated patients that were TEADA+ at any time was 11.4% compared to 3.7% of patients treated with placebo (Table 4). When considering the data of the entire treatment and post-treatment study period (weeks 4–24), the frequency of TEADA+ at any time increased to 30.6% (81/265) for galcanezumab (Table 4, no placebo data were available). Among them, 51 patients (63%) first became TEADA+ at week 24 (approximately 16 weeks after the last dose of galcanezumab), and 61 patients (75.3%) had a maximum titer of 1:40 or less; maximum titers of 1:80, 1:160, 1:320 and 1:640 were reported by twelve, four, two and two patients, respectively.
None of the patients who reported an SAE or discontinued due to an AE were TEADA+ at any time during the study. Overall, TEAEs were reported at similar frequencies in galcanezumab-treated patients with status of TEADA+ (46.9%) and TEADA− (55.4%). All the TEADA+ patients with an injection site event became TEADA+ after the event was reported and dosing had completed. Similarly, all TEADA+ patients with a hypersensitivity event became TEADA+ for the first time at week 24 after the event was reported.
Impact of immunogenicity on pharmacokinetics and pharmacodynamics
The concentrations of galcanezumab and CGRP, the target ligand for the galcanezumab antibody, were assessed at weeks 4, 12, 18, and 24 in galcanezumab-treated patients (across all doses) classified as TEADA+ and TEADA−. The concentrations of galcanezumab (Online Supplementary Information S3) and CGRP (Online Supplementary Information S4) were generally similar in patients who were TEADA+ and TEADA− when concentrations were compared at the same time point within a dose level. To note, the majority of patients that were TEADA+ at any time also tested positive in the neutralizing assay (96.3% over the 24-week period).
Vital signs and weight
During treatment and post-treatment periods, mean changes in systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse were not clinically meaningful, and there were no trends to show that galcanezumab treatment increased blood pressure. However, at some individual visits, statistically significant differences were observed in individual galcanezumab dose groups compared with placebo (Figure 2).
Change from baseline in sitting systolic blood pressure, diastolic blood pressure, and pulse using mixed model repeated measures (MMRM) analysis. Results are least squares (LS) mean changes from baseline in placebo-treated patients and galcanezumab-treated patients (all dose groups) during the treatment period (weeks 0–12) and the post-treatment period (weeks 12–24). *p-value ≤ 0.05 for the dose group compared with placebo at the indicated individual visit.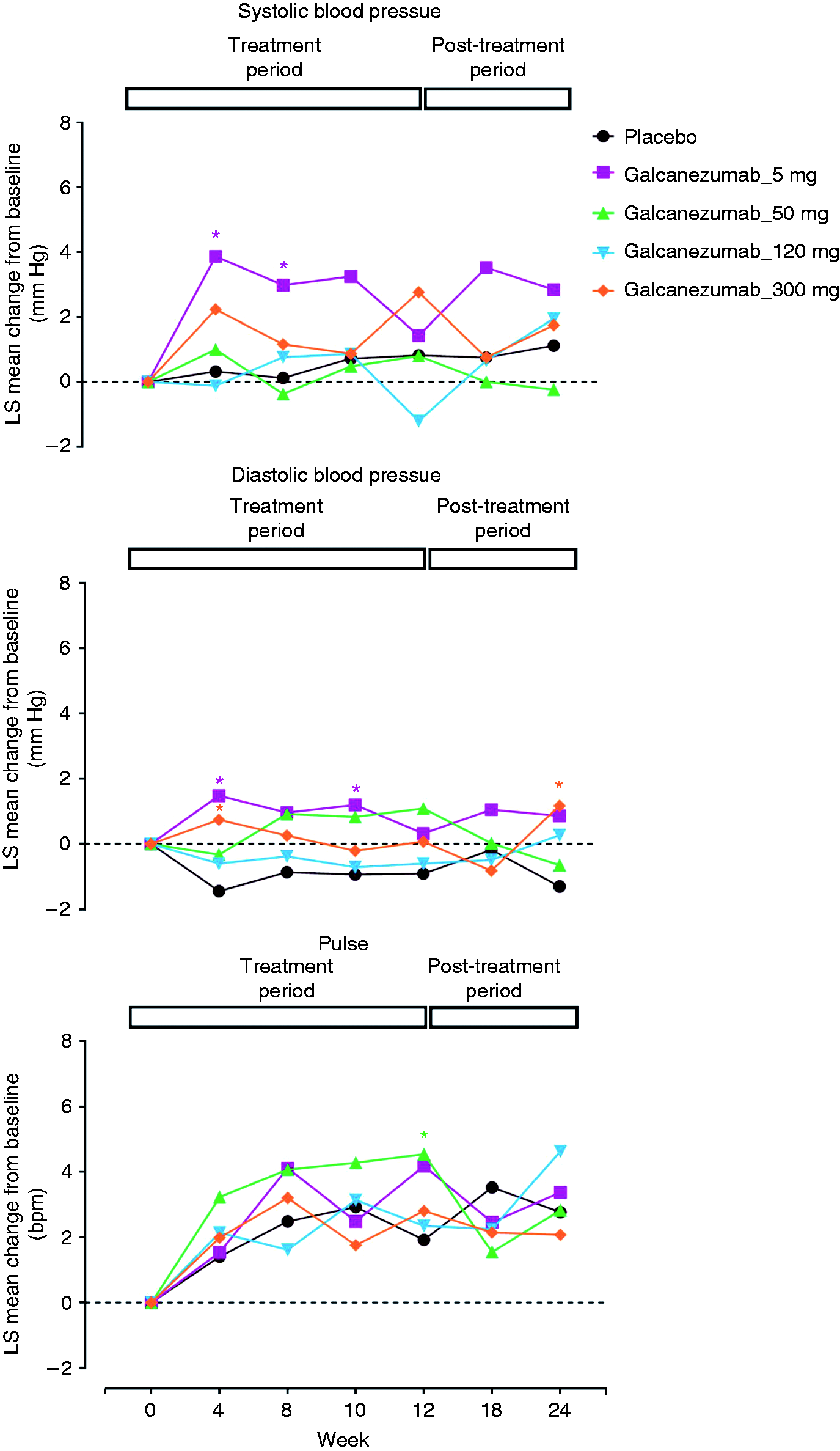
Incidence of treatment-emergent abnormal changes in vital signs was low and not significantly different between galcanezumab_all and placebo groups: high SBP (≥140 mm Hg and increase ≥20 mm Hg) (galcanezumab_all 0.4%; placebo 0%), high DBP (≥90 mm Hg and increase ≥10 mm Hg) (galcanezumab_all 1.1%; placebo 2.2%) and high pulse (>100 bpm and increase ≥15 bpm) (galcanezumab_all 2.6%; placebo 2.2%). No patient experienced sustained increases in blood pressure or pulse during either the treatment or post-treatment period. During both treatment and post-treatment periods, changes in temperature were small and clinically not meaningful; weight changes were also similar across treatment groups (not shown).
Electrocardiograms
During the treatment and post-treatment period, mean baseline-to-endpoint changes in ECG intervals (PR, QRS, and QTcF) and heart rate showed no clinically meaningful differences between individual or pooled galcanezumab dose groups and placebo; only one dose of galcanezumab (50 mg) showed a statistically significant mean decrease (−1.96 ms) in QRS interval relative to placebo during the treatment period, which was not considered clinically significant. None of the changes in ECG intervals were dose proportional. There were also no clinically meaningful or significant differences in treatment-emergent abnormal changes in heart rate, PR, QRS, and QTcF intervals at any time between any individual dose or galcanezumab_all dose group, and placebo.
Discussion
In this Phase 2b study in patients with episodic migraine, a large proportion of galcanezumab-treated patients completed the study (87%), and there were low frequencies of discontinuation due to an AE (galcanezumab_all: Treatment period 0.7%; post-treatment period, 0.4%). The majority of TEAEs in the current study were rated as mild to moderate, and similar numbers of galcanezumab- and placebo-treated patients reported at least one TEAE (51%). The frequency and severity of TEAEs with galcanezumab treatment are similar to those reported for other antibodies against the CGRP pathway in Phase 2 studies (16–18).
The AE profile reported in this study is consistent with a peripheral site of action for galcanezumab. The common TEAEs in this study were treatment-emergent injection-site pain, URTI and nasopharyngitis, of which injection-site pain was the only TEAE that was reported by statistically significantly more patients in the galcanezumab_all group compared with placebo (Table 2). Adverse events related to the injection site demonstrated a dose-dependent response. (Table 4). During a Phase 2 study with one antibody against CGRP (TEV-48125), injection-site pain was the most commonly reported AE (18). URTI and nasopharyngitis were the most frequently reported AEs in Phase 2 studies of two other antibodies targeting the CGRP pathway, ALD403 and AMG334, respectively. URTI was reported by 9% and 7% of patients treated with ALD403 and placebo, while nasopharyngitis was reported in 9%, 5%, 6% and 8% of patients treated with AMG334 7, 21 or 70 mg, and placebo, respectively (16,17). However, a recent meta-analysis of four randomized Phase 2 studies with four different antibodies against the CGRP pathway (TEV-48125, ALD403, AMG334 and galcanezumab) showed no difference in incidence of URTI or sinusitis between the study drug and placebo groups (19).
For patients who reported potential hypersensitivity events (galcanezumab_all 3.3%), none were severe, none were symptoms of anaphylaxis and none led to discontinuation. Changes in laboratory values were minimal and did not appear to be clinically important, with mean values remaining well within normal range. Further, increases in hepatic enzyme levels, which had led to termination of development of telcagepant, a small molecule inhibitor of the CGRP receptor (20), were not observed in this study with galcanezumab. CGRP is a potent vasodilator (5); however, inhibition of this effect of CGRP with galcanezumab did not lead to clinically significant changes in vital signs or ECGs in this study.
The incidence of TEADAs during the treatment period (weeks 4–12) was 4.6% in the galcanezumab_all group. None of the patients who were TEADA+ in the study reported an SAE or discontinued due to an AE. There was no data to suggest a meaningful correlation between ADA status and TEAEs.
Galcanezumab and CGRP concentrations were similar in the patients who were TEADA+ and TEADA−, suggesting that immunogenicity does not alter the pharmacokinetics of galcanezumab or its ability to bind to the target ligand. Of note, the majority of CGRP is likely bound to galcanezumab at the doses administered in this study. Since the clearance of the galcanezumab-bound CGRP is much slower than the clearance of free CGRP, the concentration of CGRP increases following galcanezumab administration. If immunogenicity had an effect on galcanezumab binding to CGRP, then lower concentrations of CGRP may have been observed in TEADA+ patients. It is well documented that ligand-antibody complexes often take on disposition characteristics of the antibody and not the unbound ligand (21).
These Phase 2b safety results are similar to those observed in a previous Phase 2a clinical study (13), with the most common TEAE for galcanezumab being injection site pain. The frequency of discontinuation for any reason was low and TEAEs were predominately rated as mild to moderate. Limitations of this study include the relatively short duration, and exclusion of patients with significant medical illness, active psychiatric disease, other types of headache and clinically significant laboratory abnormalities. The data from this Phase 2b study suggest that galcanezumab is well tolerated, with a safety profile similar to placebo with respect to changes in laboratory analytes, vital signs, and ECGs. Results support the development of galcanezumab in larger Phase 3 studies of longer duration.
Clinical implications
In this Phase 2b study in patients with episodic migraine, the adverse event profile of galcanezumab was consistent with a peripheral site of action. Similar numbers of galcanezumab- and placebo-treated patients reported at least one TEAE and less than 1% of patients discontinued due to an adverse event. There was no evidence of drug-induced hepatotoxicity and there were no clinically meaningful changes in laboratory parameters. Treatment with galcanezumab did not result in sustained increases in mean blood pressure, pulse, or ECG parameters. Development of anti-drug antibodies following galcanezumab treatment did not lead to any clinically meaningful effects on safety, pharmacokinetics, or pharmacodynamics.
Footnotes
Acknowledgements
Sriram Govindan, a full-time employee of Eli Lilly, provided medical writing assistance for this manuscript. Tonya Quinlan, a full-time employee of Eli Lilly, performed analysis of the pharmacokinetic and pharmacodyamic data.
Trial registration
This study is registered with ClinicalTrials.gov, number NCT02163993.
Sponsor
This study was supported by Eli Lilly and Company, Indianapolis, IN 46285, USA.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Tina Marie Myers Oakes, Vladimir Skljarevski, Qi Zhang, William Kielbasa, Michael Hodsdon and Holland C Detke are employees of Eli Lilly and Company, Indianapolis, IN, USA and hold company stock. Angelo Camporeale is an employee of Eli Lilly Italia and holds company stocks. Joel R Saper reports research grants/consulting fee/honoraria from Alder, Allergan, Amgen, Autonomic Technologies, Colucid Pharm, Daiichi Sankyo, Dr. Reddy’s Laboratories, Eli Lilly, Scion, Supernus Pharm, Teva, Tonix, Zosano and Migraine Research Foundation.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.