
Abstract
Background
Recent genome-wide association studies have identified transient receptor potential M8 (TRPM8) as a migraine susceptibility gene. TRPM8 is a nonselective cation channel that mediates cool perception. However, its precise role in migraine pathophysiology is elusive. Transient receptor potential V1 (TRPV1) is a nonselective cation channel activated by noxious heat. Both TRPM8 and TRPV1 are expressed in trigeminal ganglion (TG) neurons.
Methods
We investigated the functional roles of TRPM8 and TRPV1 in a meningeal inflammation-based migraine model by measuring the effects of facial TRPM8 activation on thermal allodynia and assessing receptor coexpression changes in TG neurons. We performed retrograde tracer labeling to identify TG neurons innervating the face and dura.
Results
We found that pharmacological TRPM8 activation reversed the meningeal inflammation-induced lowering of the facial heat pain threshold, an effect abolished by genetic ablation of TRPM8. No significant changes in the heat pain threshold were seen in sham-operated animals. Meningeal inflammation caused dynamic alterations in TRPM8/TRPV1 coexpression patterns in TG neurons, and colocalization was most pronounced when the ameliorating effect of TRPM8 activation on thermal allodynia was maximal. Our tracer assay disclosed the presence of dura-innervating TG neurons sending collaterals to the face. Approximately half of them were TRPV1-positive. We also demonstrated functional inhibition of TRPV1 by TRPM8 in a cell-based assay using c-Jun N-terminal kinase phosphorylation as a surrogate marker.
Conclusions
Our findings provide a plausible mechanism to explain how facial TRPM8 activation can relieve migraine by suppressing TRPV1 activity. Facial TRPM8 appears to be a promising therapeutic target for migraine.
Keywords
Introduction
Migraine-related disabilities are frequently uncontrollable, mainly because current migraine management remains suboptimal. Traditionally, cooling of craniofacial structures has been used to relieve migraine headaches. The perception of cool temperatures (<23℃) is mediated by transient receptor potential M8 (TRPM8), a nonselective cation channel activated by cooling agents such as menthol and icilin (1–4). TRPM8 is abundantly expressed in trigeminal ganglion (TG) neurons (1,2). Intriguingly, a randomized placebo-controlled study demonstrated that menthol application to the face was effective in aborting migraine attacks (5), suggesting TRPM8 activation as a possible therapeutic strategy for migraine.
A widely-accepted mechanism posits that migraine headache is caused by activation of the trigeminovascular system, including TG neurons innervating the dura mater, where calcitonin gene-related peptide (CGRP) acts as an important neurotransmitter (6,7). The tenet that the disease locus responsible for migraine headache lies in the peripheral nervous system is consistent with the recently reported efficacy of monoclonal antibodies against CGRP or its receptor in migraine prophylaxis (8–11). Moreover, several genome-wide association studies (GWASs) have identified TRPM8 as a candidate susceptibility gene for migraine (12–14). Nevertheless, the precise pathogenic role of TRPM8 remains unknown, although as stated above, TRPM8 activation is likely protective against migraine (5).
Another member of the TRP family, transient receptor potential V1 (TRPV1), is a nonselective cation channel that is activated by noxious stimuli such as high temperatures (>43℃) and capsaicin stimulation (15). TRPV1 colocalizes with CGRP in nociceptive TG neurons. The cation channel is also implicated in migraine pathophysiology. When activated, TRPV1 promotes CGRP release from trigeminal terminals (16). Moreover, a recent study reported increased TRPV1 expression in the trigeminal fibers of chronic migraine patients (17).
The meningeal inflammation induced by inflammatory soup (IS) is known to cause a transient sensitization of the dural trigeminal system (18) and is used as a migraine model in rodents (19–21). We found that IS-induced meningeal inflammation lowered the threshold temperature for heat pain withdrawal of the face. Pharmacological activation of TRPM8 with icilin reversed this thermally sensitized state, an action that was abrogated by genetic deletion of TRPM8. In parallel, IS-induced meningeal inflammation caused dynamic changes in the expression of TRPM8 and TRPV1 in TG neurons, accompanied by increased channel colocalization. Our retrograde tracer assay identified TG neurons innervating both the dura and the face. Although these neurons were found in the ophthalmic (V1) and maxillary (V2) divisions of the TG, the former segment was found to harbor a significantly larger number of such neurons. We also demonstrated cell-autonomous functional inhibition of TRPV1 by TRPM8 in a cell culture system. These findings provide invaluable insights into the role of TRPM8 in migraine pathophysiology and could lead to the development of novel TRPM8-based therapeutic strategies.
Materials and methods
Animals
Male C57BL/6 mice (CLEA Japan Inc., N = 66, age 10−12 weeks, 20−25 g) and TRPM8 knockout (KO) mice (Jackson Laboratory, Bar Harbor, ME, N = 24, age 12–16 weeks, 22−27 g) were used in this study. They were housed in cages with free access to water and food. Three animals were used for a dual retrograde tracer assay, nine animals for in situ hybridization, 30 animals for immunohistochemistry, and the remaining animals for behavioral analysis of facial heat pain. All experimental procedures were approved by the Laboratory Animal Care and Use Committee of Keio University (Authorization No. 14005), and all studies were conducted in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
IS-induced meningeal inflammation model
Mice were anesthetized with isoflurane (1.0% in room air) at 37℃. We installed a small open cranial window 2 mm in diameter centered at bregma. After the dura mater was exposed, inflammation was induced by locally applying 5 µl of IS (1 mM each of histamine, serotonin, and bradykinin and 0.1 mM prostaglandin E2 in 10 mM HEPES buffer, pH 5.5) (20). The application site was then covered with the skull bone and dental cement. As we used the small amount of IS, and the overlying skull bone was already denervated, concern for spread of IS to the surrounding tissue and stimulation of periosteal trigeminal endings was minimal. The mice were sacrificed six hours, 24 hours (Day 1), 48 hours (Day 2), or six days (Day 6) after inflammation induction. Sham-operated mice underwent the same craniotomy but no IS treatment, and were sacrificed six days later. Control animals did not undergo any surgical procedure or IS treatment.
Behavioral heat pain test
Before surgery (described above), mice were pretrained until they were consistently able to stay calm in an experimental apparatus that restricted body mobility except for head movement. On the day before experimental data collection, successfully pretrained mice were anesthetized with 1% isoflurane, and facial hair was removed. The next day, a pair of Peltier module bars with surface temperature regulated between 36℃ and 56℃ was applied to the face bilaterally. The bars were in contact with the bilateral periorbital regions and whiskers. The bar surface temperature was gradually elevated from 36℃ by 1℃/4 seconds until face withdrawal, a behavior index of thermal nociception. Mouse behaviors were monitored by a video recorder (Panasonic, Kadoma Japan). Video analysis was performed by an examiner blind to the identity of the animals. The lowest temperature at which a mouse averted the head away from the bars was considered the heat pain threshold temperature for the animal. In each session, measurement of the threshold temperature was repeated five times. After baseline thresholds were collected, the mice were subjected to sham operation or IS-induced meningeal inflammation as described. A 5 mm × 5 mm piece of filter paper immersed in either icilin (10 mM) or vehicle (dimethyl sulfoxide: DMSO) was applied to the face on each side for five min. This treatment was carried out 30 min prior to each behavioral test. We remeasured threshold temperatures at six hours, 24 hours (Day 1), 48 hours (Day 2), and six days (Day 6) after IS administration. As for the sample size calculation for the behavioral study, our preliminary experiments revealed that the standard deviation (SD) of the heat pain threshold temperature of untreated control mice was 0.5℃. With the type I error rate and power being 5% and 0.80, respectively, if we were to detect a 0.5–1.0℃ difference, the sample size required was calculated as 4–16. Accordingly, we used six animals and measured the threshold temperature in pentaplicate at every measuring time point, as stated above.
Immunostaining and in situ hybridization
TG tissue was prepared as described elsewhere (22,23). Serial sections of 10 µm thickness were prepared for histological examination. Sections were immunostained with rabbit anti-TRPM8 (KM060, TransGenic Inc., Kobe, Japan) and goat anti-TRPV1 (sc-12498, Santa Cruz Biotechnology, Dallas, TX). Immunoreactivity was visualized using species-specific donkey secondary antibodies conjugated to Cy3 or fluorescein isothiocyanate (FITC) (Jackson ImmunoResearch Labs, West Grove, PA). We also immunostained tissue sections obtained from TRPM8 KO mice with the TRPM8 antibody to check its specificity. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI: Sigma-Aldrich, St. Louis, MO). The immunolabeled specimens were examined under a Keyence BIOREVO BZ-9000 microscope (Keyence, Osaka, Japan) and a TCS-SP5 confocal laser scanning microscope (Leica Microsystems, Mannheim, Germany). For cell counting, we counted TRPM8-positive and TRPV1-positive cells and calculated the ratio of each to all DAPI-positive neurons. We also calculated the proportion of TRPM8-positive cells in the entire TRPV1-positive cell population. We conducted in situ hybridization for TRPM8 mRNA according to a protocol described elsewhere (23). The probe sequences were designed for nucleotides 279−978 of the mouse TRPM8 cDNA sequence (GenBank Accession No.: AF481480).
Identification of TG neurons innervating the dura and face by retrograde tracers
To confirm the existence of TG neurons innervating both the dura and face, under anesthesia with 1–2% isoflurane, the retrograde axonal tracers Fluorogold (FG; Biotium, Hayward, CA) and DiI (Life Technologies, Carlsbad, CA) were applied to the dura and subcutaneous tissue of both whisker pads of untreated wild-type mice, respectively. For FG administration, a round piece (2 mm in diameter) of the skull bone at bregma was removed. Care was taken not to damage the underlying dura. FG (approximately 100 µg) was put evenly over the surface of the exposed dura. The skull bone piece was returned, and the overlying skin was sutured. Meanwhile, DiI solution (20 mg/ml, 50 µl) was injected into the subcutaneous tissue of the bilateral periorbital regions and whisker pads. After recovery from anesthesia, the animals were kept individually with free access to food and water. Three days after tracer application, the animals (N = 3) were sacrificed and transcardially perfused with 4% paraformaldehyde/phosphate-buffered saline. The bilateral TGs were dissected out, and 10 µm thick TG tissue sections were prepared on a cryostat. For tissue sections containing the V1 and V2 divisions of the TG, the numbers of tracer-labeled cells were counted by three independent examiners. We performed TRPV1 immunostaining for a subset of sections prepared from these TG samples in the same protocol described above.
Stable transformants expressing an emerald GFP (EmGFP)-rat full-length TRPM8-V5 epitope fusion protein
Total RNA was prepared from the TG of an adult male Sprague-Dawley rat using TRIZOL LS Reagent (Life Technologies). cDNA was synthesized using the SuperScript III First-Strand Synthesis System (Life Technologies). Full-length TRPM8 cDNA was amplified by PCR using a set of sequence-specific primers (forward: 5’-caccatggccttcgagggagccagg-3’, reverse: 5’-tttgactttattagcaatctctttcag-3’). The amplified DNA fragment was subcloned into pcDNA™3.2-DEST (Life Technologies). The EmGFP-rat full-length TRPM8-V5 expression vector was transfected into PC12 cells using Lipofectamine 2000 (Life Technologies). Clones of stably transfected cells were isolated using 10 µg/ml Blasticidin (Life Technologies). All experimental procedures were approved by Keio University School of Medicine Safety Committee on Genetically Modified Organisms (Authorization No. 20-017-5).
Calcium imaging
EmGFP-rat full-length TRPM8-V5-expressing PC12 cells were incubated with 5 µM Rhod-2 AM (Thermo Fisher Scientific, Waltham, MA) in imaging solution containing 117 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgSO4, 25 mM HEPES, and 30 mM D-(+)-glucose, (pH 7.4) at 37℃ for 20 min, followed by washing for 30 min in the imaging solution. For image capture, the cells were perfused at 10 ml/min with the imaging solution at room temperature and then exposed to the imaging solution, containing varying concentrations of icilin. Images were acquired at 2 Hz (500 ms exposure time) with a cooled CCD camera (Andor iXon, DU897) connected to a Nikon Eclipse microscope with a 20 × (NA 0.45) objective lens. Imaging analysis was performed with ImageJ software (NIH).
Western blot analysis
Western blotting of cell lysates was conducted as described (22). Blotting membranes were serially incubated with primary antibodies and species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch Labs). Immunoreactivity was visualized using Western Lightning® Plus-ECL, Enhanced Chemiluminescence Substrate (Perkin Elmer, Waltham, MA). The primary antibodies used were as follows: Anti-V5 (46-0705, Life Technologies), anti-phospho-c-Jun N-terminal kinase (JNK) (4668, Cell Signaling Technology, Danvers, MA), and anti-total JNK (9252, Cell Signaling Technology). Densitometric analysis of immunoreactive bands was conducted using Multigauge software v. 3.3 (Fuji Film, Tokyo, Japan).
Statistical analysis
All quantitative data on pain threshold temperature and band intensities are expressed as the mean ± SD. Levene’s test was used to assess the equality of variances for numerical data across groups. In the behavioral test, group means were first compared by two-way analysis of variance (ANOVA) to check whether there was any significant interaction between independent variables, followed by multiple comparison with Bonferroni’s post hoc test. For cell counting, we used a chi-square test for comparison. For analysis of western blot data, target protein band intensities were normalized to that of the corresponding internal control and expressed as the fold change from baseline. Statistical analysis was performed by one-way ANOVA followed by Bonferroni’s post hoc test or unpaired t-test. All statistical analyses were performed using IBM SPSS, v. 23 (Chicago, IL, USA), and the statistical significance was set at p < 0.05.
Results
Effects of TRPM8 stimulation on the heat pain threshold in a mouse meningeal inflammation model
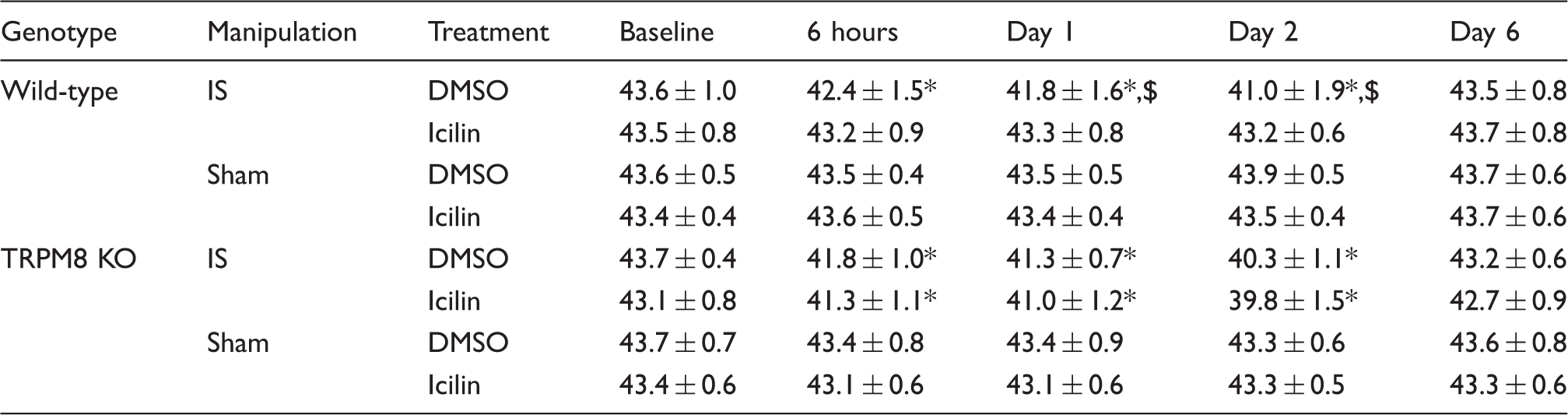
Under inflammatory conditions, TRPV1 activity at nociceptors is enhanced, causing thermal hyperalgesia/allodynia (24). Such TRPV1 hyperactivity can be assessed by a reduction in the heat pain threshold (24,25). After IS was administered to the dura in vehicle-treated wild-type mice, the heat pain threshold was significantly lower at six hours than at baseline (42.4 ± 1.5℃ vs. 43.6 ± 1.0℃ at baseline, p < 0.001, ANOVA, N = 30 at each time point, Figure 1(a) and Table 1). The threshold temperature further decreased, reaching a peak (lowest threshold temperature) on Day 2 post-treatment (41.0 ± 1.9℃, p < 0.001 vs. baseline, ANOVA, N = 30 at each time point, Figure 1(a) and Table 1). The heat pain threshold returned to the pretreatment level by Day 6 (43.5 ± 0.8℃, N = 30, Figure 1(a) and Table 1). Icilin is an established TRPM8 agonist (3,26). Compared to vehicle-treated mice, icilin-treated mice exhibited significantly higher threshold temperatures at Day 1 and Day 2 (43.3 ± 0.8℃ vs. 41.8 ± 1.6℃ at Day 1, 43.2 ± 0.6℃ vs. 41.0 ± 1.9℃ at Day 2, p < 0.001, ANOVA, N = 30 each, Figure 1(a) and Table 1). In sham-operated wild-type mice, icilin pretreatment did not have any effect on the heat threshold temperature (Figure 1(b) and Tabl 1). The inhibitory effect of icilin was not observed in TRPM8 KO mice (Figure 1(c) and Table 1). The sham operation did not alter the threshold temperature throughout the examination period in either vehicle-treated or icilin-treated TRPM8 KO mice (Figure 1(d) and Table 1). Our two-way ANOVA revealed that there was no significant effect of mouse genotype on the development of meningeal inflammation-induced heat pain threshold changes (F value = 1.414, p = 0.235).
Temporal profiles of the heat pain threshold temperatures after the induction of IS-induced meningeal inflammation. (a) IS-induced meningeal inflammation lowered the heat pain threshold temperature at six hours and Days 1 and 2 in vehicle (DMSO)-treated wild-type mice (filled triangle, N = 30 from six independent experiments). In contrast, there was no change in threshold temperature when compared to that of pre-IS Baseline in icilin-pretreated WT mice (filled circle, N = 30 from six independent experiments). *p < 0.001, ANOVA, Bonferroni’s post hoc test, compared to baseline. $p < 0.001, ANOVA, Bonferroni’s post hoc test, between-group difference at the same time point. (b) In sham-operated wild-type mice, there were no temporal changes in the heat pain threshold temperature. Icilin did not affect the heat pain threshold temperature. (N = 30 from six independent experiments in each group). (c) In TRPM8 KO mice, there were no antinociceptive effects of icilin on IS-induced threshold temperature changes (N = 30 from six independent experiments in each group). (d) In sham-operated TRPM8 KO mice, the heat pain threshold temperature did not change throughout the experimental period. There was no significant effect of icilin on the heat pain threshold temperature. (N = 30 from six independent experiments in each group). Quantitative data of thermal pain threshold changes. mean ± SD (℃). p < 0.001, vs. Baseline, ANOVA, Bonferoni's post-hoc test. p < 0.001, vs. icilin-treated, ANOVA, Bonferoni's post-hoc test.
TRPM8 and TRPV1 expression changes in TG neurons after IS-induced meningeal inflammation
Next, we sought to determine the changes in the TRPM8 and TRPV1 expression levels in TG neurons after IS-induced meningeal inflammation by immunohistochemistry and in situ hybridization. In control and sham-operated mice, immunostaining of TG sections revealed that TRPV1 and TRPM8 proteins were expressed in small- to medium-sized TG neurons, and only a small number of cells were double-positive for both TRPs (Figure 2(a), left column, arrowheads). After the induction of meningeal inflammation, however, the numbers of TG neurons expressed in either or both TRPs increased over the next two days before returning to baseline by Day 6 (Figure 2(a) and (b)). We carried out immunostaining for TG tissue sections obtained from TRPM8 KO mice with the same TRPM8 antibody. There was no significant immunoreactivity in these samples (Figure 2(c)). In situ hybridization indicated that IS-induced meningeal inflammation upregulated TRPM8 transcriptional activity (Figure 2(d)).
Changes in TRPM8 and TRPV1 expression in TG neurons during IS-induced meningeal inflammation. (a) Photomicrographs showing TRPM8-positive and TRPV1-positive TG neurons. TG neurons double positive for TRPM8 and TRPV1 are indicated by arrowheads. Bar = 15 µm. (b) Left: The ratios of TRPM8-positive (white) and TRPV1-positive (black) neurons to total neurons are shown in the bar graph. **p < 0.001, compared to control, ANOVA, Bonferroni post hoc test. Right: The colocalization ratio of TRPM8 and TRPV1 in TG neurons is shown in the bar graph. **p < 0.001, compared to control, ANOVA, Bonferroni post hoc test. (c) There was no significant immunoreactivity in TRPM8 KO mouse TG tissue with the same TRPM8 antibody. Bar = 20 µm. (d) TG neurons positive for TRPM8 transcripts visualized by in situ hybridization. For simplicity, only data for Sham (sham-operated animals) and Day 2 and Day 6 post-IS are displayed. Bar = 30 µm.
Labeling of TG neurons after retrograde tracer administration at the dura and face
The increased colocalization of both TRPs in TG neurons during meningeal inflammation suggests that these receptor channels interact to modulate TG neuron activity and sensory pain thresholds. To address whether interventions at different trigeminal innervation sites can interact within the same TG neuron, we performed a dual axonal tracer labeling study by applying FG and DiI to the dura and face, respectively (Figure 3(a)). Because dural afferents are known to project to the V1 and V2 divisions of the TG (27,28), and the receptive fields subjected to thermal stimulation corresponded to the V1 and V2 areas in our behavioral test, we conducted quantitative analyses on TG neurons in the V1 and V2 divisions. The proportion of FG-labeled TG neurons receiving dural afferents was higher in the V1 division than in the V2 division (14.3% vs. 7.7%, p < 0.001, chi-square test, Figure 3(b)). The proportion of TRPV1-positive neurons was higher in the V2 division than in the V1 division (15.5% vs. 12.1%, p < 0.001, chi-square test, Figure 3(b)). The proportion of TG neurons dually innervating the dura and face in all of the face-innervating TG neurons was higher in the V1 division (47.4% vs. 23.5%, p < 0.001, chi-square test, Figure 3(b)). The proportions of TG neurons dually innervating the dura and face in all the face-innervating TG neurons were equivalent between the two divisions (52.8% and 52.7% in V1 and V1, respectively, p = 0.519, chi-square test, Figure 3(b)). Approximately half of these dually innervating TG neurons were TRPV1-positive (41.0% and 52.3% in V1 and V2, respectively, p = 0.024, chi-square test, Figure 3(b)).
Labeling of TG neurons after retrograde tracer administration at the dura and face. (a) Identification of TG neurons innervating the dura (FG labeling) and/or face (DiI labeling). TRPV1-expressing cells double positive for FG and DiI are indicated by an arrowhead. A TG neuron innervating only the face is indicated by an arrow. TRPV1-negative TG neurons innervating only the dura (#) and a TRPV1-negative TG neuron dually innervating the dura and face (*) are indicated. Bar: 10 µm. (b) Quantitative data for retrograde axonal labeling and TRPV1 immunostaining in TG neurons are presented. The data were analyzed by chi-square test.
Functional interactions between TRPM8 and TPPV1 in cultured cells
In light of the histological results showing elevated TRPM8/TRPV1 coexpression during TG inflammation (and thermal allodynia), we explored whether TRPM8 activity antagonized TRPV1-mediated functions within identical cells. We first made an expression construct for an EmGFP-rat full-length TRPM8-V5 fusion protein (Figure 4(a)) to transfect PC12 cells. EmGFP-positive PC12 cells exhibited a steeper icilin-induced intracellular calcium rise at higher EmGFP (TRPM8) expression levels, validating the functionality of our expression construct (Figure 4(b)). Subsequently, we prepared PC12 cells stably expressing the rat full-length TRPV1 protein. We previously demonstrated that the TRPV1 agonist capsaicin increases JNK phosphorylation via TRPV1 in a dose-dependent manner (22). Hence, we used JNK phosphorylation as a surrogate marker of TRPV1 activation. These cells were then transfected with the EmGFP-TRPM8-V5 expression cassette to achieve coexpression of TRPV1 and TRPM8. We then assessed the interaction between receptor molecules by measuring JNK phosphorylation as a surrogate index of TRPV1 activity (Figure 4(c), upper row). Concomitant treatment with icilin significantly suppressed capsaicin-induced JNK phosphorylation in the presence of TRPM8 expression (Figure 4(c)).
Functional interactions between TRPM8 and TRPV1 in PC12 cells. (a) The expression construct for the EmGFP-rat full-length TRPM8-V5 epitope fusion protein. Expression was driven by the CMV promoter. (b) Calcium imaging with Rhod-2 AM revealed that the intracellular calcium increase induced by icilin was dependent on the TRPM8 expression level, such that a higher calcium rise was observed at lower icilin concentrations in high TRPM8-expressing cells. The regions of calcium concentration measurement are encircled. Low, Intermediate, and High indicate the expression levels of TRPM8 (EmGFP). Bar: 10 µm. (c) Upper row: Dose and time-dependent increases in JNK phosphorylation evoked by capsaicin (Caps) in rat full-length TRPV1-stably expressing PC12 cells. Lower row: In the absence of TRPM8 expression (left), icilin treatment did not significantly change the magnitude of TRPV1-induced JNK phosphorylation. In the presence of TRPM8 expression, icilin treatment attenuated TRPV1-induced JNK phosphorylation. A densitometric analysis of western blots is presented in the bar graph. Ratios of capsaicin-induced JNK phosphorylation in icilin-treated cells to that in vehicle-treated cells are compared between TRPM8-null and TRPM8-expressing cells. **p = 0.004, unpaired t-test. Cells were treated with 50 µM capsaicin for 60 min. Where noted, cells were pretreated 30 min prior with 100 nM icilin.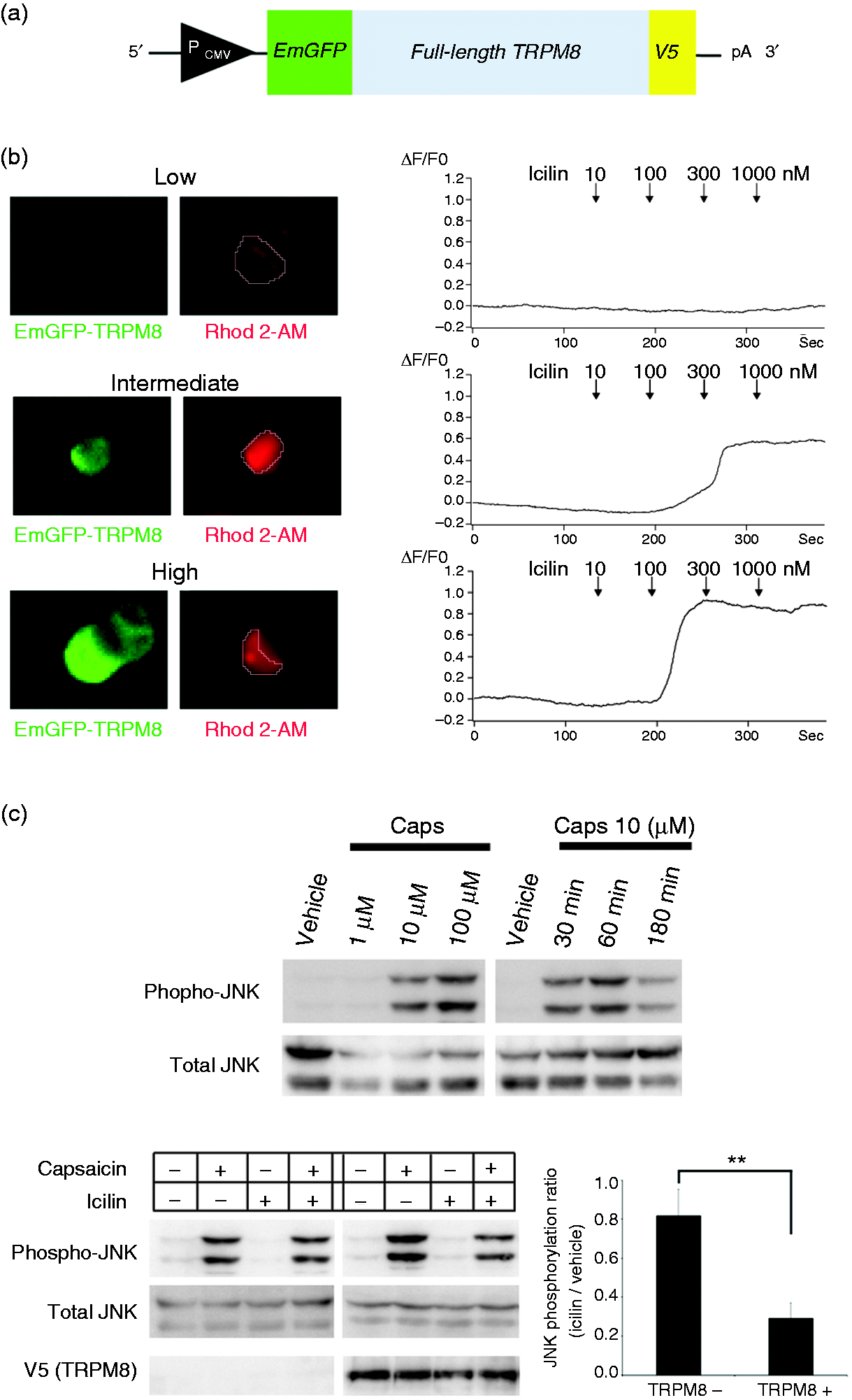
Discussion
Stimulation of TRPM8 reversed the thermal allodynia associated with IS-induced meningeal inflammation. The TRPM8-mediated antinociceptive action was dependent on the presence of meningeal inflammation because TRPM8 stimulation did not elevate the heat pain threshold temperature in sham-operated animals. This finding suggested that meningeal inflammation gave rise to a situation that enabled TRPM8 to interact with TRPV1. Consistently, IS-induced meningeal inflammation increased the proportion of TRPM8-positive neurons in the TG by transcriptional upregulation, and there was a concomitant increase in the colocalization of TRPM8 with TRPV1. Retrograde axonal tracer labeling disclosed the presence of dura-innervating TG neurons that sent collaterals to the face as well, and approximately half of these TG neurons were TRPV1-positive. Furthermore, our cell experiments showed that TRPM8 stimulation attenuated TRPV1-induced phosphorylation of JNK, implying that TRPM8 can antagonize TRPV1 function in a cell-autonomous manner. Collectively, our data suggest that facial TRPM8 activation is a promising therapeutic intervention for controlling TRPV1 activity of dura-innervating TG neurons, which is likely to have important relevance to migraine therapy.
Although the origin of migraine headache is still a matter of controversy (29), recent success in migraine prophylaxis with antibodies against CGRP or its receptor strongly supports the role of peripheral CGRP-positive trigeminal terminals in the dura (8–11). CGRP is thought to induce degranulation of mast cells in the dura, which contributes to the development of inflammation (6,30). It follows that such inflammation sensitizes the trigeminal system, and, consequently, normally innocuous cranial vascular pulsations become perceivable as throbbing pain during migraine attacks (7). IS-induced meningeal inflammation has been used as a classic animal model of migraine (20,21). Electrophysiological studies by Burstein et al. (20) demonstrated that TG neurons became sensitized to mechanical and thermal stimulation to the face at 20 min after topical IS treatment to the dura. Their subsequent study showed that the electrical sensitization of TG neurons to thermal stimulation persisted for at least two hours after IS treatment (21). In their study, the cutaneous receptive fields innervated by sensitized TG neurons included the whisker pads as well as the periorbital regions (20,21). Wieseler et al. (19) also demonstrated development of mechanical allodynia by performing von Frey filament testing in a similar IS-induced meningeal inflammation model. Furthermore, our behavioral data delineated the temporal profile of facial thermal allodynia, as seen by a lowered threshold temperature for head withdrawal, in a more extended time window. Facial thermal allodynia was most marked at Day 2, but had resolved by Day 6 after IS-induced meningeal inflammation. These experimental data indicate that an intracranial inflammatory event is capable of inducing extracranial altered sensory functions. In the classic view, such a phenomenon should be explained by sensory integration at the level of the brainstem, and development of extracranial allodynia/hyperalgesia is interpreted as an indication of central sensitization (31,32). However, recent evidence has raised the possibility that sensory input from intracranial and extracranial locations can converge at the level of TG neurons. Kosaras et al. (33) identified abundant nerve fibers along the sutures, some of which appeared to emerge from the dura. Schueler et al. (34) observed that dextran amines applied to the periosteum labeled the dura, TG, and spinal trigeminal nucleus. In agreement with this histological observation, their electrophysiological recordings revealed afferent fibers with mechanosensitive receptive fields both in the dura and in the parietal periosteum (34). Our retrograde axonal tracer study has provided further anatomical evidence for sensory integration at the level of the TG neurons. Our observation that the V1 division exhibited a higher proportion of dually innervating neurons of the entire population of dural afferent neurons was consistent with previous reports (27,28).
TRPV1 is known to be implicated in inflammation-related sensitization to thermal stimulation. Genetic deletion of TRPV1 conferred complete resistance to carrageenan-induced thermal hyperalgesia in mice (25). The pivotal role of TRPV1 in inflammation-induced thermal hyperalgesia/allodynia has been substantiated by other studies (35–40). Regarding the relationship between TRPV1 and TRPM8, there are human studies showing that TRPM8 agonists, such as menthol (41) and peppermint oil (42), attenuate TRPV1-mediated pain in the trigeminal territory, although the precise mechanism underlying such antinociceptive actions remains obscure. There have been several reports on the coexistence of TRPV1 and TRPM8 in individual TG neurons (43–45). In the present study, we found that TRPM8 expression was increased in TG neurons after IS-induced meningeal inflammation through transcriptional upregulation. As a result, the number of TRPM8/TRPV1-positive TG neurons was increased, and the most pronounced colocalization of both TRP channels was observed with the greatest efficacy of icilin for relieving thermal allodynia. These findings support the view that the analgesic action of icilin is exerted at the level of primary sensory neurons (TG neurons) via TRPM8.
Our statistical analysis showed that genetic ablation of TRPM8 itself did not affect the trajectory of heat pain threshold alterations after IS-mediated meningeal inflammation. However, we found a trend indicating that icilin treatment led to a non-significant but lower heat pain threshold temperature throughout the examination period in IS-mediated meningeal inflammation-subjected TRPM8 KO mice (Figure 3(c) and Table 1). This raises the possibility that icilin can cause heat hyperalgesia/allodynia through its TRPM8-independent action(s). TRPM8 modulators have been reported to be able to cause altered body temperature and paradoxical temperature sensation (46–48). These facts should be kept in mind with attempts to use TRPM8 modulators, including icilin, in clinical practice.
We previously demonstrated that JNK phosphorylation can serve as a surrogate marker of TRPV1 activity in our cell system (22). In the present study, icilin pretreatment was observed to reduce TRPV1-mediated phosphorylation of JNK only in the presence of heterologous TRPM8 expression. To the best of our knowledge, such a functional interaction between TRPM8 and TRPV1 in a cell-autonomous manner has been demonstrated only in colonic sensory neurons (49). How can facial TRPM8 activation alleviate the thermal allodynia induced by meningeal inflammation in a cell autonomous manner? In the basal condition, there are only a small number of TRPM8/TRPV1-positive TG neurons (Figure 5(a)). Meningeal inflammation activates TRPV1-positive dura afferent TG neurons. After meningeal inflammation, TRPM8 expression is gradually upregulated through transcriptional activation, which leads to increased coexpression of TRPM8 and TRPV1. Some of these TRPM8/TRPV1-positive neurons innervate the dura and face (Figure 5(b) and (c)). In this state, facial TRPM8 stimulation can reverse TRPV1-mediated thermal allodynia in a cell-autonomous manner (Figure 5(d)).
Proposed mechanism by which facial TRPM8 activation alleviates meningeal inflammation-mediated thermal allodynia. (a) In the resting state, there are few TG neurons that express both TRPV1 and TRPM8. Some of the dural afferent TG neurons send collaterals to the face as well. (b) Meningeal inflammation can activate TRPV1-positive TG neurons, which causes headache and facial thermal allodynia. (c) After a while, TRPM8 expression is enhanced by transcriptional upregulation. As a consequence, the number of TRPM8/TRPV1-positive TG neurons increases. Such TRPM8 upregulation in TRPV1-positive cells also occurs in TG neurons innervating both the dura and face. (d) In this condition, facial TRPM8 activation can alleviate TRPV1-mediated thermal allodynia and, possibly, headache. In this paradigm, opposing actions derived from the intracranial (dura) and extracranial (facial tissue) tissue can interact with each other in a cell-autonomous fashion.
There are several limitations to our study. Expansion of the receptive field has been recognized as an important feature of IS-induced facial thermal allodynia (21). Unfortunately, our experimental device for facial heat pain testing was not suitable for spatial assessment of receptive fields. Moreover, histological analysis of dural tissue after IS-induced inflammation was impossible in our experimental model because of the considerable adhesion between the skull and dura after IS administration. We previously reported that TRPV1-positive nerve fibers are abundant in the dura (50). Meanwhile, there is a controversy concerning dural innervation of TRPM8-positive fibers. Local icilin administration to the dura caused cutaneous allodynia in rats (51), indicating that the dura was responsive to TRPM8 stimulation. However, a previous study using transgenic mice expressing farnesylated enhanced GFP from one TRPM8 allele demonstrated that dural TRPM8-positive nerve fibers were scarce in adulthood owing to postnatal fiber pruning (52). Our finding implies that TRPM8 expression might be enhanced by local inflammation in the meningeal nerve terminals as well as in TG neurons. However, we were unable to clarify this point. In addition, we did not address any central action of TRPM8 in the present study. Our data do not exclude the coexistence of any central mechanisms with respect to the antinociceptive effect of facial TRPM8 stimulation. As for cell-based experiments, we should have ideally used primary TG neuron-rich cultures. That may have rendered our study much more relevant to the actual clinical setting. Capsaicin concentrations required for JNK phosphorylation in our cells (22) and CGRP release in primary TG neurons (53) seem to differ from each other. However, in the primary culture system, the number of obtained viable TG neurons is not so high that biochemical analysis using western blotting would be almost impossible. Instead, by using PC12 cells, which derive from the neural crest like TG neurons, we were able to obtain biochemical data steadily. Importantly, the TRPV1 expression level in our PC12 cells was not so high, because we used a stable TRPV1-expressing cell line (22).
In summary, our results strongly suggest that facial TRPM8 activation can exert an antimigraine action by inactivating TRPV1 function at the level of TG neurons. While these findings may provide important insights into migraine pathophysiology, it should be noted that TRPM8 and TRPV1 are also involved in the pathophysiology of other craniofacial disorders, such as meningitis, so the applicability of our results may be extensive.
Article highlights
TRPM8 activation can exert an analgesic action by antagonizing TRPV1 at the level of TG neurons. Meningeal inflammation upregulates TRPM8 expression in TG neurons by enhancing transcriptional activity. Facial TRPM8 activation is a promising therapeutic intervention for migraine.
Footnotes
Acknowledgements
We are grateful to the Collaborative Research Resources of Keio University School of Medicine for equipment use.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by JSPS KAKENHI (Grant numbers 26460706 to MS), a grant from the Takeda Science Foundation to MS, and research grants from Pfizer Inc. (WS1878886) and Nippon Zoki Pharmaceutical Co., Ltd.to NS.