
Abstract
Background
Hemicrania continua (HC) is a primary headache syndrome characterized by a unilateral, moderate, continuous headache with exacerbations marked by migrainous and cranial autonomic symptoms. However, clinical phenotypes similar to primary HC may be subtended by several disorders.
Case report
We report the case of a 62-year-old man experiencing, over the previous year, a headache completely consistent with HC and its absolute responsiveness to indomethacin therapy. Later, the patient developed diplopia caused by sixth cranial nerve palsy ipsilateral to headache. In this frame, clinical, laboratory and neuroimaging characteristics supported the diagnosis of idiopathic hypertrophic pachymeningitis (IHP).
Conclusions
IHP is a rare fibrosing inflammatory disorder leading to a localized or diffuse dura mater thickening. IHP clinical manifestations are a progressively worsening headache and signs related to cranial nerves involvement and venous sinus thrombosis. Here, we report, for the first time, a HC phenotype subtended by IHP.
Keywords
Introduction
Hemicrania continua (HC) is a syndrome characterized by a unilateral, moderate, fluctuating, continuous headache, which is relatively featureless except during exacerbations when both migrainous and cranial autonomic symptoms (such as tearing, conjunctival injection and ptosis ipsilateral to the pain) may be present (1). More specifically, the clinical picture is characterized by an enduring pain (present 24 hours a day, seven days a week, for at least three months) with transient worsening, ranging from minutes (sometimes even less than five minutes) to days or even weeks (2). During these exacerbations, migraine features, such as light sensitivity ipsilateral to the side of the pain, noise intolerance and nausea, can be present (2). Several other clinical characteristics not included in the HC criteria of the third edition of the International Classification of Headache Disorders (ICHD), beta version (3), have also been observed in this syndrome, including an itching eye (many patients report a foreign body sensation, like an eyelash or a piece of sand in the eye ipsilateral to the pain) and facial or cheek swelling (4). Although the diagnosis of HC is based on clinical history and neurological examination, its hallmark is the absolute response to indomethacin according to the International Headache Society criteria (3). However, misdiagnosis in HC is frequent (5) and likely due to its relative rarity, autonomic signs paucity and distinct similarities to migraine during exacerbations. In this regard, it is crucial to consider that a clinical phenotype similar to primary HC may be subtended by other neurological, neoplastic or systemic disorders (6). Among these, unruptured intracranial aneurysms, internal carotid artery dissection, non-metastatic and metastatic lung cancer, inflammatory orbital pseudotumor, nasopharyngeal carcinoma, intracranial tumors and HIV infection have been extensively reported. Furthermore, although several cases of trigeminal-autonomic cephalalgias related to meningeal disorders, such as viral meningitis or pachymeningitis, have been previously described (7), to the best of our knowledge, none of these presented with HC clinical features.
Case report
We report the case of a 62-year-old man admitted to hospital owing to 12 months of constant moderate pain, with throbbing exacerbations of about two to three hours strictly localized in the left orbital and temporal regions. During these episodes, conjunctival injection, tearing and mild ptosis occurred, although relatively mildly, in various combinations, ipsilateral to the pain. Physical and neurological examinations and a brain magnetic resonance imaging (MRI) scan without gadolinium were normal whereas a laboratory test showed high levels of inflammatory markers (erythrocyte sedimentation rate (ESR): 60 mm/L/hour).
Considering the patient’s age, the pain features and an ESR of more than 50 mm/L/hour, a first diagnostic hypothesis of giant cell arteritis was proposed and both a steroid therapy (dexamethasone) and an appropriate diagnostic process were started. Despite the clinical improvement, the headache and accompanying autonomic signs not only had never gone completely away but significantly upsurged concomitantly with the corticosteroid tapering.
For this reason, after about eight weeks the patient was referred to the Headache Center of the University Hospital of Naples where the atypical clinical picture, in the absence of temporal artery thickening and hardening, jaw claudication, visual acuity abnormalities and negativity of diagnostic tests (the ESR was normal in this phase), suggested that the hypothesis of giant cell arteritis was unlikely. Contrariwise, persistent headache with concomitant migrainous and trigeminal-autonomic symptoms during exacerbations and a dramatic clinical response to the intramuscular administration of 50 mg indomethacin (in the so-called Indotest) (8) suggested the alternative diagnosis of HC and the consequent oral indomethacin treatment (50 mg three times daily). Four weeks later, the patient returned to our observation due to the abrupt occurrence of diplopia. The Hess Lancaster screen test revealed the sixth cranial nerve palsy ipsilateral to previous symptoms. In this context, a fundus oculi examination and visual potential were performed and did not show abnormalities. A further brain MRI scan with gadolinium revealed widespread but asymmetrical pachymeningeal thickening (more prominent in the left side) and left transverse and sigmoid cerebral sinus thrombosis (Figure 1). Altogether, the clinical picture and laboratory and neuroimaging findings supported the diagnosis of hypertrophic pachymeningitis (9). Therefore, additional tests to exclude secondary forms of hypertrophic pachymeningitis were performed. Blood evaluation, serum angiotensin converting enzyme, c-ANCA and p-ANCA, rheumatoid factor, antinuclear antibody, analysis for Lyme, syphilis, tuberculosis, and fungi infection, chest X-ray, 18F-FDG PET/CT and cerebrospinal fluid (CSF) analysis for infection and neoplasia were unremarkable. However, the ESR value was 99 mm/L/hour and CSF lymphocytosis with high protein levels (suggesting blood–brain barrier damage likely due to concomitant leptomeningeal involvement) was observed (9). Finally, plasma and CSF immunoglobulin subtype dosage, the IgG4-index and the IgG4-Loc allowed us to exclude IgG4-related pachymeningitis (10). During the lumbar puncture, as routinely done, the opening CSF pressure was evaluated (14 mm H2O). Consequently, the diagnosis of idiopathic hypertrophic pachymeningitis (IHP) was considered (9).
Magnetic resonance imaging (MRI) sequences pre (A–D) and post (A’–D’) therapy and both before (A–A’) and after (B–D and B’–D’) intravenous contrast agent (gadolinium) administration. A and A’ images revealed marked dural thickening, diffusely involving the left tentorium, with a characteristic low T2 signal intensity. In this context, both the physiologic flow void as well as the slow flow phenomena are absent in the homolateral sigmoid sinus. After the gadolinium contrast examination (B–D) intense and uniform enhancement of the mastoid petroclival and tentorial dural thickening is observed. This MRI finding is present also in the left middle cranial fossa, along the temporal lobe, spreading to the left cavernous sinus and involving the homolateral dura of the trigeminal mandibular branch (V3), markedly enlarged (arrow in C and C’) compared to the right (arrowhead in C and C’). Post-therapy MRI (A’–D’) revealed a decrease of dural thickening without changes to the slow flow phenomena at the level of the sigmoid sinus.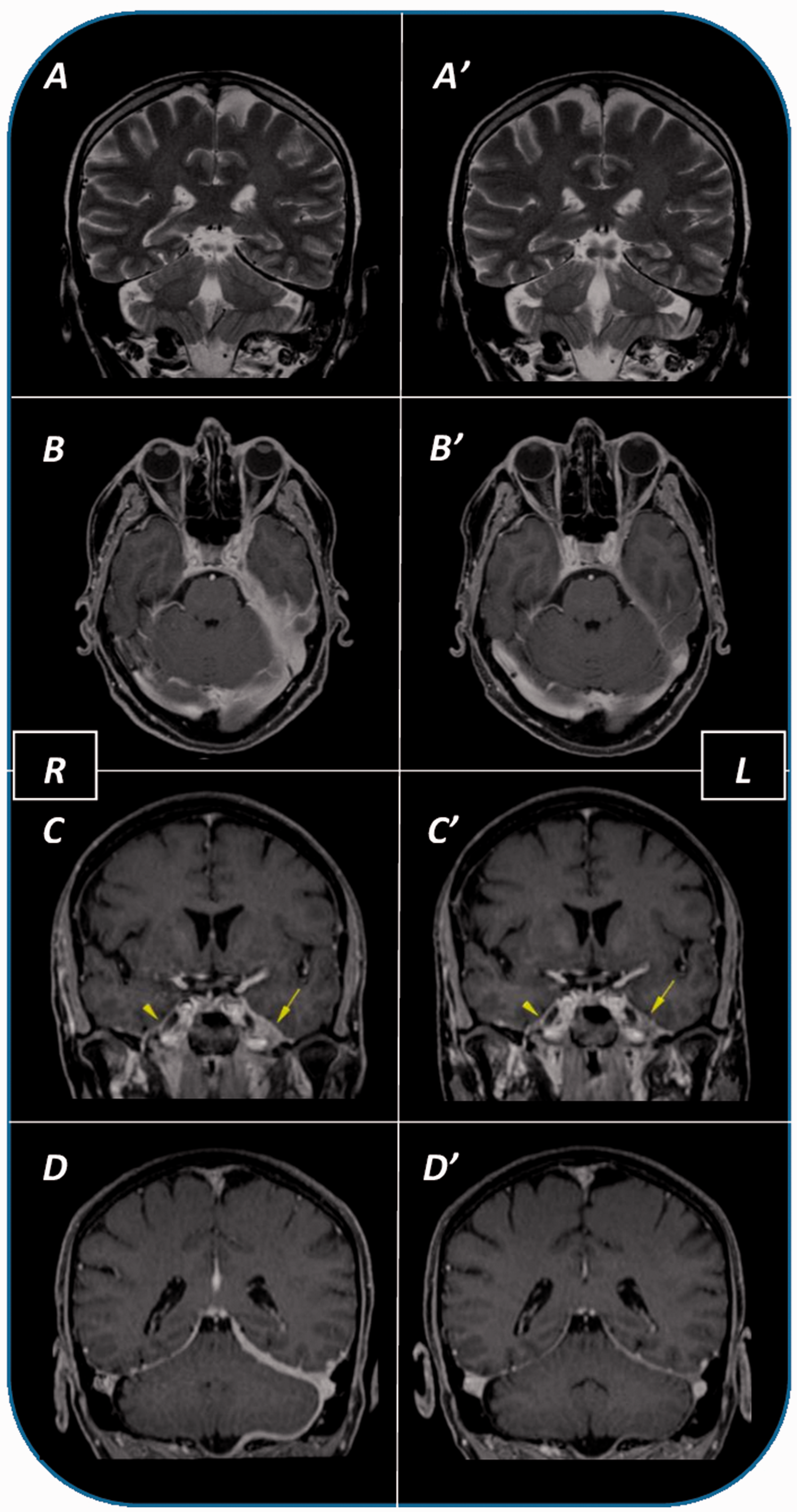
Thus, a therapy with both methylprednisolone (50 mg daily) and enoxaparin sodium (100 IU/kg b.i.d.) was suddenly started. After three days, we observed the resolution of the sixth nerve palsy as well as the remission of the continuous baseline pain and the superimposed exacerbations. After 15 days, a follow-up brain MRI scan with gadolinium depicted minimal pachymeningeal thickening of the clivus, making it arduous to proceed with the planned meningeal biopsy.
Currently, the patient is following a combination therapy with both oral methylprednisolone (25 mg daily) and azathioprine (125 mg daily) and is exhibiting a very good clinical response.
Discussion
IHP is a rare fibrosing inflammatory disorder leading to a localized or diffuse cranial or spinal dura mater thickening, which usually affects patients over 60 years old, with a higher prevalence in males (9).
The first IHP clinical manifestation is usually a progressively worsening headache (occurring in 90% of patients) (9). Although the IHP-related headache features are non-specific, we report an IHP exhibiting a peculiar HC phenotype, in which left pain and autonomic signs were respectively due to ipsilateral pachymeningeal inflammation and fibrous encasement of trigeminal nerves (Figure 1). Other common disease manifestations are related to involvement of cranial nerves (11); specifically, the more often affected are the optic nerves, with deficits both in visual acuity and in visual field, and the oculomotor nerves, with diplopia and ophthalmoplegia (11). Venous sinus thrombosis is another common manifestation, as well as ataxia and hyperprolactinemia, respectively due to cerebellar and pituitary compression (9,11). The increase of inflammation markers, although poorly specific, is particularly sensitive and useful because its value, being directly related to disease severity, is an index of response to therapy.
Neuroimaging investigation allows the completion of IHP diagnosis by showing the meningeal thickening associated with high-contrast enhancement. Nevertheless, several pathological entities, potentially able to induce the dura mater enhancement (such as the intracranial hypotension due to spontaneous or traumatic cerebrospinal fluid leak), must be excluded before concluding for an idiopathic form. Similarly, meningeal disorders presenting as a thickening and inflammatory process of dura mater have been described in relation to specific infections (including Lyme disease, syphilis, Mycobacterium tuberculosis, fungal infections, cysticercosis, HTLV-1, and malignant external necrotizing otitis due to Pseudomonas) and autoimmune disorders (including Wegener granulomatosis, rheumatoid arthritis, sarcoidosis, Behçet disease, Sjögren syndrome, and temporal arteritis). Moreover, hypertrophic pachymeningitis can be related also to systemic or local neoplastic disorders, such as carcinomatosis, lymphoma, and meningioma en plaque. Furthermore, IgG4-related IHP, a recently described entity, is an increasingly recognized manifestation of IgG4-related disease, a fibro-inflammatory condition that can affect virtually any organ (10). It is estimated that IgG4-RHP may account for a high proportion of cases of hypertrophic pachymeningitis once considered idiopathic (12). Finally, the absence of both symptoms consistent with widespread systemic (e.g. fever, rash, musculoskeletal symptoms) and organ-specific (e.g. recurrent ocular manifestations, sensorineural hearing loss) inflammation and the normal values of inflammatory markers (e.g. C-reactive protein and serum amyloid A) excluded the diagnosis of autoinflammatory syndromes like TNF receptor (TRAPS) or cryopyrin-associated periodic (CAPS) syndromes, despite the aseptic meningitis and ESR increase.
IHP treatment is often challenging. On one hand, a brilliant response to steroid therapy is typical and should be considered an auxiliary diagnostic criterion in doubtful cases. On the other hand, both the many potential adverse effects associated with long-term steroid therapy and the disease recurrences (frequently observed during the drug tapering) often make it necessary to introduce, over time, a steroid-sparing therapy such as azathioprine, methotrexate, cyclosporine or mycophenolate (9).
In conclusion, to the best of our knowledge, this is the first case in which a headache fulfilling ICHD-III beta criteria for HC represents the clinical presentation of IHP. Therefore, we encourage the attention of clinicians in considering a suitable laboratory and neuroimaging diagnostic process to rule out putative secondary HC, also in the presence of a typical phenotype.
Clinical implications
HC is a rare primary headache syndrome but a phenotype mimicking HC could be subtended by several diseases. In our patient, a headache completely consistent with HC, also with absolute responsiveness to indomethacin therapy, is secondary to idiopathic hypertrophic pachymeningitis (IHP). The IHP treatment is often challenging: nevertheless, a brilliant response to steroid therapy is typical and should be considered an auxiliary diagnostic criterion in doubtful cases. Consequent steroid-sparing therapies such as azathioprine, methotrexate, cyclosporine or mycophenolate should be considered. Laboratory and neuroimaging diagnostic examinations to rule out secondary HC are mandatory, also in the presence of a typical HC phenotype.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.