
Abstract
Aim
To provide an overview of mechanisms underlying craniofacial pain; to highlight peripheral and central adaptations that may promote chronification of pain in craniofacial pain states such as migraine and temporomandibular disorders (TMD).
Background
Pain is a common symptom associated with disorders involving craniofacial tissues including the teeth and their supporting structures, the temporomandibular joint and the muscles of the head. Most acute painful craniofacial conditions are easily recognized and well managed, but others, especially those that are chronic (e.g., migraine, TMD and trigeminal neuropathies), present clinical challenges. Preclinical studies have provided substantial information about the anatomical and physiological mechanisms related to the initiation and modulation of nociceptive signals in the trigeminal system. While knowledge of the mechanisms underlying chronic craniofacial pain remains limited, both clinical and preclinical investigations suggest that changes in afferent inputs to the brain as well as in brain structure and modulatory pathways occur in chronic pain. Collectively, these changes result in amplification of nociception that promotes and sustains craniofacial chronic pain states.
Conclusions
The increased understanding gained of the physiological and pathological processing of nociception in the trigeminal system has provided new perspectives for the mechanistic understanding of acute craniofacial pain conditions and the peripheral and central adaptations that are related to pain chronification. Such knowledge may contribute to improvements in currently available treatments as well as to the development of novel analgesic therapies.
Keywords
Abbreviations
serotonin brain-derived neurotrophic factor calcitonin gene-related peptide central nervous system Cycloxygenase conditioned pain modulation diffuse noxious inhibitory controls protooncogene product FOS gamma amino butyric acid low-threshold mechanosensory medication overuse headache N-methyl-D-aspartate nociceptive-specific periaqueductal grey matter Prostaglandin E2 post-traumatic trigeminal neuropathy rostroventral medial medulla substance P traumatic brain injuries temporomandibular disorders temporomandibular joint transient receptor potential trigeminal brainstem sensory nuclear complex wide dynamic range
Introduction
The craniofacial region comprises a wide variety of tissues and structures that include the facial skin, meninges, oral mucosa, teeth and their surrounding periodontal tissues, periosteum, bone, temporomandibular joint (TMJ), muscles, ligaments, fascia, etc. The face and mouth also have special psychological and emotional meaning and importance in a variety of functions including eating, drinking, speech and expression of emotions as well as several sensory functions, some of which are unique to this part of the body (e.g, taste, smell). The craniofacial region also has a rich innervation and an extensive somatosensory representation in the central nervous system (CNS). This region is also the site of some of the most common acute and chronic pain conditions. These include temporomandibular disorders (TMD) and several types of toothaches, headaches, and trigeminal neuropathic pain conditions. Most acute painful craniofacial conditions are easily recognized and can be well managed, but other conditions, especially those that are chronic, are more challenging to manage effectively. This chapter provides an overview of recent advances in the understanding of the mechanisms within the craniofacial tissues and the CNS that are integral for the initiation, processing and modulation of the neural signals related to acute and chronic craniofacial pain conditions. These insights may contribute to improvements in therapeutic approaches that are currently available, as well as to the development of novel analgesic therapies. More detailed outlines and original source references related to these mechanisms can be found in (1–6).
Craniofacial pain processing
Peripheral nociceptive mechanisms
The craniofacial tissues are innervated almost exclusively by branches of the trigeminal (V) sensory nerve (although some small areas of the craniofacial region are supplied by other cranial nerves or cervical nerves), and the cell bodies of the V primary afferent nerve fibers are predominantly located in the V ganglion. The ophthalmic branch, or first division, of the V nerve mainly supplies the supraorbital tissues (e.g., forehead skin), meninges and cornea, its maxillary branch, or second division, principally innervates the infraorbital skin, upper lip, maxillary mucosa and teeth, and the mandibular branch, or third division, supplies mainly the skin of the lower jaw, lower lip, mandibular mucosa and teeth. The V primary afferent fibers terminate in these tissues as sense organs (receptors), many of which are quite complex in their structure and are activated by tactile or proprioceptive stimuli. These low-threshold mechanoreceptors are mainly associated with large (A-beta) or medium (A-delta)-sized primary afferent fibers. Other primary afferents may terminate as free nerve endings, many of which are activated by noxious stimuli and function as nociceptors. These nociceptive primary afferents are either small-diameter myelinated (A-delta) fibers, or even smaller and slower-conducting unmyelinated (C) fibers; A-beta afferents may take on a nociceptive function in some pain conditions. Not all of the A-delta and C-fiber afferents, however, carry nociceptive information, since some are associated with receptors that respond to non-noxious cooling, warming or even tactile stimuli (2,4,6).
The activation of the different types of afferent endings may result in the firing of action potentials in their associated nerve fibers that are then conducted into the CNS. These inputs provide sensory-discriminative information about the location, quality, intensity, and duration of the stimulus that activates the endings, and in the case of nociceptors, results also in affective-motivational qualities of pain. Nociceptors may also develop a prolonged increase in excitability after tissue injury or inflammation, so much so that they may develop ongoing (i.e., “spontaneous”) activity and become more responsive to noxious stimulation or even begin to respond to stimuli that are normally innocuous (2,3). This process, termed “peripheral sensitization” may contribute to the increased sensitivity of an injured or inflamed tissue.
The activation or peripheral sensitization of the nociceptive endings involves many different types of chemical mediators (Figure 1). These include several mediators that also have actions in the CNS (e.g. glutamate, gamma amino butyric acid [GABA], serotonin [5-HT] and noradrenaline as well as neuropeptides); and there may be sex differences in the peripheral actions of some of these mediators (2,5,6). It is also noteworthy that the nociceptive afferents can undergo phenotypic adaptations or even “switches” under certain conditions (see (2,3,7) for review). For example, in response to peripheral inflammation, the expression of certain receptors or ion channels (eg, sodium channels) may be altered. Transcription of ion channels, neuropeptides and brain-derived neurotrophic factor, and translation of TRP channels may occur and enhance peripheral sensitization (Figure 1). Transcriptional changes may also occur after nerve injury and contribute to the development of abnormal, ectopic discharge patterns in the afferents that are often a feature of neuropathic pain states such as trigeminal neuralgia (3,5,8–11). These changes occurring in inflammatory or neuropathic conditions may be manifested in the neuronal cell bodies in the V ganglion, as well as in the V afferent fibers themselves, and involve a number of chemical mediators as well as the satellite glial cells that encapsulate the ganglion cell bodies (2,3,5,6). The heightened excitability of the afferents may contribute to the spontaneous nature, allodynia, hyperalgesia (see below) as well as extra-territorial spread often seen in inflammatory or neuropathic pain states (3,5).
Mediators in craniofacial tissues that are involved in peripheral sensitization following inflammation. As part of the inflammatory process, numerous chemicals are released from mast cells, immune cells, macrophages, and injured cells that act on ion channels or membrane receptors on peripheral nociceptive afferent nerve endings and thereby may alter the sensitivity of the endings. Several of the mediators are shown. Some of these mediators produce an increase in the excitability of the nociceptive afferent endings (i.e., peripheral sensitization), and others may exert inhibitory effects. ASIC, acid-sensing ion channel; CRH, corticotrophin-releasing hormone; GIRK, G-protein-coupled inward rectifying potassium channel; 5-HT, serotonin; iGluR, ionotropic glutamate receptor; IL-1b, interleukin-1-beta; IL-6, interleukin-6; LIF, leukemia inhibitory factor; μ, μ-opioid receptor; M2, muscarinic receptor; mGluR, metabotropic glutamate receptor; NGF, nerve growth factor; PAF, platelet-activating factor; PGE2, prostaglandin E2; PKA, protein kinase A; PKC, protein kinase C; SSTR2A, somatostatin receptor 2A; TNF-α, tumor necrosis factor alpha; TrkA, tyrosine kinase receptor A; TRPV1, transient receptor potential vanilloid 1; TTXr, tetrodotoxin-resistant sodium channel (Reproduced with permission from Meyer et al. (119)).
There are some notable differences between V and spinal nociceptive afferents in these peripheral changes. These include the apparent lack of sympathetic efferent sprouting in the V ganglion after V nerve injury, in contrast to the sympathetic efferent sprouting seen in dorsal root ganglia after spinal nerve injury. In addition, there are differences between both systems in the up or down regulation of neuropeptides and ion channel expression in the ganglion cells or their peripheral afferent terminals, as well as in the time course of the abnormal afferent discharge patterns (12–15).
Clinical implications of peripheral sensitization and related events
Peripheral sensitization of the afferents may contribute to ongoing pain, hyperalgesia (increased sensitivity to a stimulus that is normally painful) and allodynia (pain resulting from a stimulus not normally evoking pain) that can occur in many acute or chronic pain conditions. A common clinical example is the increased sensitivity of an acutely inflamed tooth to thermal stimuli (i.e. dental pulpitis pain).
Also of clinical significance is the multiplicity of peripheral chemical mediators, receptors, ion channels and intracellular processes involved in the activation or sensitization of the nociceptive afferents. These represent potential targets for the development of more effective therapeutic approaches to control pain. Indeed, improved understanding of peripheral nociception has led to the development of therapeutic agents targeting specific mechanisms. Examples include COX-2 inhibitors, which have their principal analgesic and anti-inflammatory actions in peripheral tissues (e.g., on PGE2 synthesis) by reducing inflammation, modulating nociceptive afferent excitability, and altering the hyperalgesia associated with some craniofacial pain states.
Brainstem nociceptive mechanisms
The V primary afferents project via their cell bodies in the V ganglion into the brainstem, where the vast majority synapse with second order neurons in the V brainstem sensory nuclear complex (VBSNC). The VBSNC is subdivided into the main (or principal) sensory nucleus and the spinal tract nucleus, which comprises three components: subnuclei oralis, interpolaris, and caudalis (1,5,16). Those V afferents activated by craniofacial tactile or proprioceptive craniofacial stimuli synapse on low-threshold mechanosensory (LTM) neurons at all levels of the VBSNC, including the subnucleus caudalis (Figure 2). In contrast, the V afferents activated by innocuous thermal stimuli or noxious stimuli terminate on neurons predominantly located in subnucleus caudalis or adjacent spinal dorsal horns of C1-C2. Since the subnucleus caudalis has many morphological and physiological features analogous to those of the spinal dorsal horn, subnucleus caudalis is often referred to as the medullary dorsal horn (1,2,5,14). However, subnucleus caudalis is not the only component of the VBSNC receiving nociceptive afferent inputs; many nociceptive afferents also project to the subnuclei oralis and interpolaris, and to the caudalis/interpolaris transitional zone.
Schema of major somatosensory pathway from the orofacial region. The cell bodies of most primary afferents in the trigeminal nerve are in the trigeminal ganglion and project to second-order neurons in the trigeminal brainstem sensory nuclear complex, which is made up of the trigeminal main sensory nucleus and the trigeminal spinal tract nucleus; the latter has three subnuclei: Oralis, interpolaris, and caudalis. These neurons may project to neurons at higher levels of the brain (e.g. in the somatosensory thalamus) or to brainstem regions such as the reticular formation or the cranial nerve motor nuclei. Not shown are the projections of some afferents in cranial nerves VII, IX, X, and XII and in cervical nerves to the trigeminal brainstem sensory nuclear complex and the projections of many V, VII, IX, and X afferents to the solitary tract nucleus (Reproduced with permission from Sessle (1)).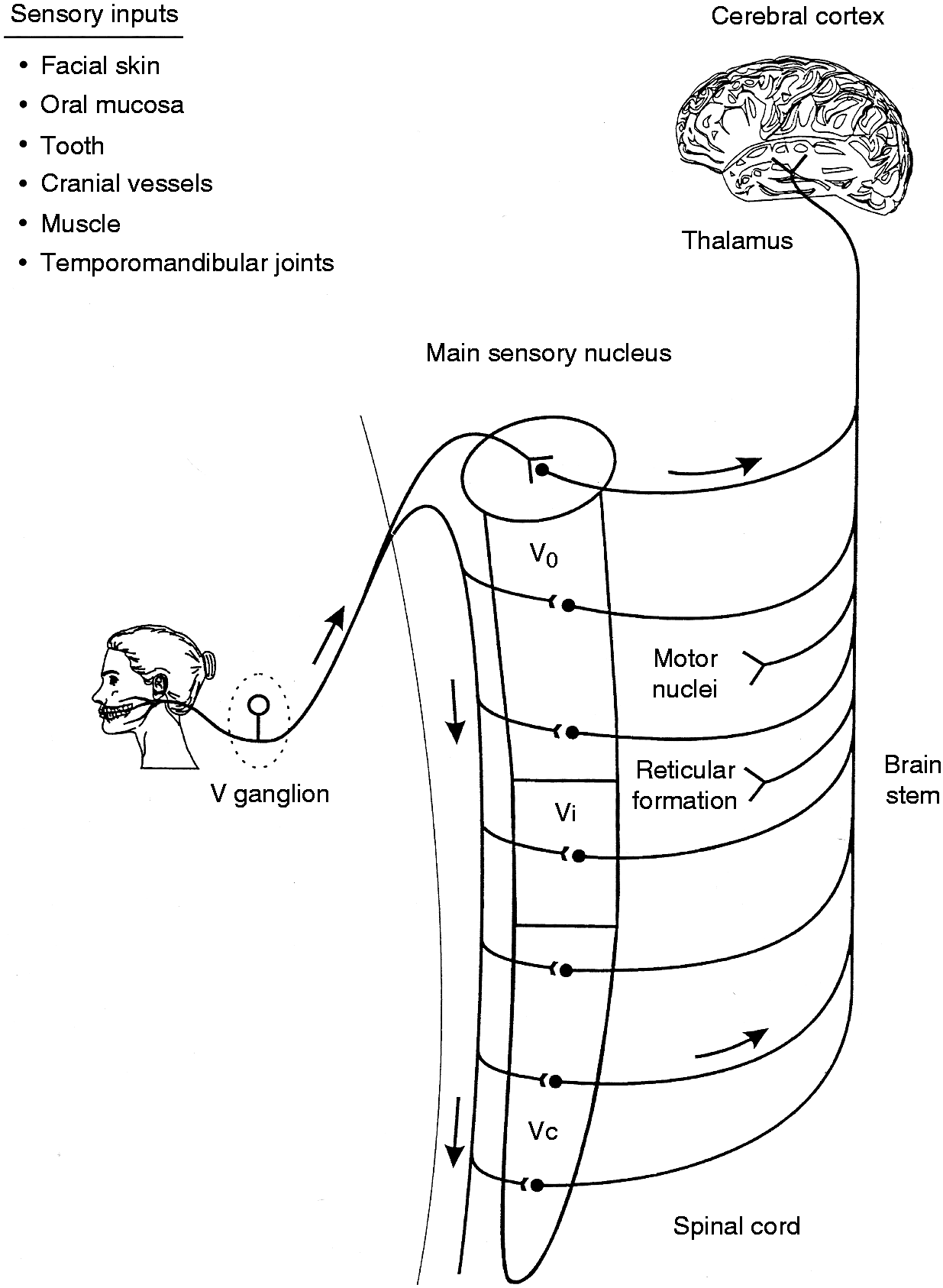
Like their spinal afferent counterparts, the central endings of A-delta and C-fiber V nociceptive afferents release several excitatory neurotransmitters or neuromodulators; these include the excitatory amino acid glutamate as well as peptides including substance P (SP) and calcitonin gene-related peptide (CGRP) (1–3,16). These chemical mediators can excite the second-order nociceptive neurons on which the afferents synapse. There are two main types of second order nociceptive neurons. The first type is the nociceptive-specific (NS) neuron that is activated only by noxious stimuli (e.g. pinching, heat) applied to the localized receptive field (e.g. on the skin) of the A-delta and C-fiber nociceptive afferents. The second type is the wide dynamic range (WDR) neuron that is activated by synaptic inputs from A-beta and A-delta afferents excited by non-noxious stimuli (e.g. tactile) as well as from A-delta and C-fiber afferents excited by noxious stimuli. Sex differences may exist in receptive field size and some of the response properties of these afferents and neurons, as well in their expression of functional receptors (17–20).
Many of the WDR and NS neurons located in subnucleus caudalis and C1-C2 dorsal horns receive their peripheral afferent inputs derived exclusively from superficial tissues (e.g. facial skin, oral mucosa). This nociceptive information is then relayed from these neurons to higher brain regions including those involved in perceptual aspects of pain and in modulatory functions (see section 3.1 below). The receptive fields and response characteristics of these neurons endow them with coding properties indicating that they are critical brainstem neural elements for the detection, localization and discrimination of superficial pain. However, nociceptive information from deep tissues (e.g., muscle, TMJ, tooth pulp, meninges) is processed predominantly by subsets of these ‘cutaneous’ nociceptive neurons that occur in subnucleus caudalis, C1-C2 dorsal horns, and the caudalis/interpolaris transition zone. In addition to their cutaneous/mucosal afferent inputs, these neurons also receive extensive convergent afferent inputs from these deep tissues as well as from cervical spinal afferents. These convergence patterns underlie the CNS processes contributing to deep pain, and in addition may explain the poor localization, spread, and referral of pain that are typical of craniofacial pain states involving deep tissues (e.g. TMD, some toothaches and headaches) and some cases of cervical pain (1,2,4,5).
The convergent afferent inputs noted above may become especially effective in activating the WDR and NS neurons in pathophysiological situations. Neuroplastic changes can be produced in the nociceptive neurons by nociceptive afferent inputs evoked in the setting of injury or inflammation (1–5, 21). This neuroplasticity is initiated by the release from the central endings of the nociceptive afferents of some of the neurochemicals mentioned above. One of these neurochemicals is glutamate, which induces a cascade of intracellular events in the WDR and NS neurons by its action on glutamatergic receptors (e.g. N-methyl-D-aspartate [NMDA]) on the nociceptive neurons. This cascade can result in neuroplastic changes reflected in an increase in neuronal excitability, termed “central sensitization” of the nociceptive neurons. Central sensitization is normally reversible in acute pain states, but, if it is maintained, it can lead to and sustain chronic pain. It underscores the point that the nociceptive pathways and processes in the CNS are not “hard-wired” but rather are “plastic” and modifiable by events associated with injury or inflammation in peripheral tissues. Recent findings in animal models of craniofacial pain have also revealed that central sensitization can involve both non-neuronal (i.e. glial) as well as ascending and descending neural processes (see below and 2–5,22). The neuroplastic changes in the nociceptive neuronal properties induced by injury or inflammation include increased receptive field size and responses to noxious stimuli and decreased activation threshold, and are associated with reflex neuromuscular changes and other behavioral alterations. These changes represent processes that, along with peripheral sensitization, can explain the hyperalgesia and allodynia as well as pain spread and referral that characterize many pain states. Central sensitization has been shown to be a feature of animal models of toothaches, headaches, TMD, and several craniofacial neuropathic pain states (see 1–6,23–27).
The axons of some neurons in the VBSNC ramify within the VBSNC and modulate the activity of other neurons in this structure (1,16). Many VBSNC neurons as well, or instead, project to other brainstem areas including the reticular formation, raphe nuclei, parabrachial area, cranial nerve motor nuclei and spinal ventral horn, and thereby contribute to the central circuitry underlying autonomic and muscle reflex responses evoked by stimulation of craniofacial tissues (1,3,5). Some of these areas are components of descending modulatory systems that influence nociceptive transmission (see section 3.1 below). Many neurons in the VBSNC also, or instead, contribute to pathways ascending in the CNS to the ipsilateral and especially contralateral thalamus (Figure 2).
Thalamocortical nociceptive mechanisms
The main thalamic areas receiving the craniofacial somatosensory information relayed from the VBSNC or other brainstem areas are the ventrobasal complex (termed the ventroposterior nucleus in the primate), the medial thalamus and the posterior nuclear group (2,5,16). LTM and thermoreceptive neurons occur in these thalamic areas, and most project to analogous neurons in the overlying somatosensory area of the cerebral cortex where their relayed signals are processed to provide for the detection, localization and discrimination of tactile and non-noxious thermal stimuli. These thalamic areas also contain WDR and NS neurons, most of which have spatiotemporal coding properties and connections to neurons in the somatosensory cerebral cortex that indicate a role for them in defining the spatiotemporal features of noxious stimuli, and thus in the sensory-discriminative dimension of pain. However, the spatiotemporal coding properties of most nociceptive neurons in the medial thalamic nuclei and posterior nuclear group and their connections, mainly to the prefrontal, anterior cingulate or insula cortical areas, suggest a role in the cognitive, motivational or affective dimensions of pain, consistent with brain imaging findings in humans (28). These areas are considered to be part of an extensive and largely distributed brain network that is often referred to as the “pain matrix”, i.e. brain areas that are generally activated during nociceptive stimulation. Higher cortical pain processing in craniofacial pain and other types of somatic pain does not appear to be fundamentally different (29,30,31,32). It is also noteworthy that neurons in these higher levels of the CNS are also subject to neuroplastic changes reflecting central sensitization following injury or inflammation in peripheral tissues.
Craniofacial pain modulation
Modulation of CNS nociceptive processes
The experience of pain is dependent upon active modulation within CNS circuits. Modulation of nociceptive inputs results from context, experience, stress, anxiety, emotional state, attention and others, contributing to the inter-individual differences in pain reported following noxious stimuli. The transmission of craniofacial somatosensory information can be modified at brainstem and thalamocortical levels (1,2,4,5,16,32). A case in point is the manifestation of central sensitization, which has been mentioned above. To varying degrees, modification may also be operational during different behavioral states, e.g. different stages of sleep and consciousness (33).
In the case of craniofacial nociceptive transmission, the variety of inputs and interconnections in V subnucleus caudalis provide an intrinsic circuitry allowing for considerable interaction between the various afferent inputs derived from peripheral tissues and from descending projections originating in several CNS areas, which include the reticular formation, raphe nuclei, parabrachial area, locus coeruleus, hypothalamus, amygdala, and several areas of the cerebral cortex (e.g. sensorimotor; anterior cingulate; prefrontal) that project to some of these subcortical areas. A number of chemical mediators, including GABA, glycine, 5-HT, noradrenaline, dopamine, orexin, and opioids (e.g., enkephalins, endorphins), provide a rich endogenous chemical substrate by which many of the primary afferent and descending inputs to the VBSNC can exert their modulatory effects on craniofacial nociceptive transmission. Inhibitory influences exerted by many of these inputs on WDR and NS neurons have been implicated as intrinsic CNS mechanisms that contribute to the analgesic effects of several therapeutic approaches used to control pain, including acupuncture, deep brain stimulation, opiate-related drugs (e.g. morphine) and antidepressants (e.g. amitriptyline, duloxetine). Some of the descending projections to the nociceptive neurons instead have a role in facilitating nociceptive transmission, such as in the central sensitization process noted earlier (4,5). Thus, descending modulation is bidirectional and can either enhance or diminish the transmission of nociceptive signals and thus influence the experience of pain (Figure 3).
Schematic representation of pain modulation circuitry. Nociceptive inputs enter the spinal dorsal horn through primary afferent fibers that synapse onto second-order transmission neurons. Ascending projections target the thalamus, and collateral projections also target mesencephalic nuclei, including the DRt, the RVM, and the midbrain PAG. Descending projections from the DRt are a critical component of the DNIC pathway. Rostral projections from the thalamus target areas that include cortical sites and the amygdala. The lateral capsular part of the CeA (“nociceptive amygdala”) receives unprocessed nociceptive inputs directly from the brainstem and spinal cord. Inputs from the thalamus and cortex enter through the lateral (LA) and basolateral (BLA) amygdala. The CeA sends outputs to cortical sites and the thalamus. Descending pain modulation is mediated through projections to the PAG, which also receives inputs from other sites, including the hypothalamus (data not shown), and communicates with the RVM as well as other medullary nuclei that send descending projections to the spinal dorsal horn through the DLF. The noradrenergic locus coeruleus (LC) receives inputs from the PAG, communicates with the RVM, and sends descending noradrenergic inhibitory projections to the medullary and spinal dorsal horns. Antinociceptive and pronociceptive spinopetal projections from the RVM positively and negatively modulate nociceptive inputs and provide for an endogenous pain regulatory system. Ascending (red) and descending (green) tracts are shown schematically. Areas labeled “i–iv” in the small diagram correspond with labeled details of the larger diagram (Reproduced with permission from Ossipov et al. (120)).
Central circuits modulating craniofacial pain
Ongoing pain, touch, and cold-induced allodynia and hyperalgesia, are especially devastating to the lives of patients with many craniofacial pain disorders including trigeminal neuralgia, painful post-traumatic trigeminal neuropathy (PTTN), TMD, oral cancer and others (34–36). Following injury, primary afferents exhibit peripheral sensitization and often display abnormal, ectopic firing that may account not only for allodynia and hyperalgesia but also for ongoing pain (see section 2.2). Central sensitization also promotes amplification of incoming signals, and central adaptative changes are reflected by neuroplastic alterations in the properties of WDR and NS neurons that likely promote pain chronification. An important mechanism of amplification is related to the descending influences on nociceptive transmission; in this case, a net increased facilitation occurring in the descending influences has been demonstrated in multiple chronic and functional pain disorders in both preclinical and human studies (4,5,37–40). In this section, we review the influence of central pain-modulatory circuits in the CNS on craniofacial pain and highlight the changes in these circuits that may contribute to chronic pain associated with different craniofacial pain disorders.
Concepts of pain have evolved from unidimensional views of a strict relationship between the degree of injury and the magnitude of pain to current understanding of the multidimensional nature of pain and its relationship to suffering; such conceptualization is encapsulated in the biopsychosocial model of pain (41,42). It is now understood that the experience of pain does not necessarily reflect the state of the body tissues and varies greatly between individuals (43). Genetic and environmental influences can modify the pain experience; prospective studies in humans, for example, have shown that genetic factors markedly influence the likelihood of an individual developing TMD (44,45). In humans, pain has long been recognized to be an experience that is the synthesis of a complex interplay of sensory, affective and cognitive features (46). The spatiotemporal features of pain are thought to be largely associated with the processing by the somatosensory cortex of the nociceptive inputs that it receives from subcortical somatosensory relays, while other cortical and limbic systems (e.g. anterior cingulated cortex, prefrontal cortex, amygdala, ventral tegmental area, nucleus accumbens) are more closely associated with encoding the affective, motivational and contextual features of pain (38, 47). Importantly, the activation of nociceptors usually elicits pain in humans, but not always (48, 49). Pain is different from nociception, in that the brain modulates nociceptive inputs to shape the human experience of pain (50).
As noted above (see section 3.1), descending modulation arises from several areas in the cerebral cortex as well from the hypothalamus, amygdala and other sites converging on the brainstem (see 38). It is noteworthy that the periaqueductal grey matter (PAG) is well positioned to modulate nociceptive inputs and pain perception through its interactions with these descending projections from higher brain areas as well as ascending projections from numerous sites. The PAG has been shown to activate an endogenous pain-inhibitory system. Microinjection of opioids into the PAG or electrical stimulation of the PAG can elicit strong opioid-sensitive antinociception in animals (38–40,51,52) and in humans (53–55). The PAG receives inputs from cortical sites and has reciprocal connections with the amygdala (56,57) as well as ascending nociceptive inputs from the spinal dorsal horn and VBSNC by way of the parabrachial nuclei (57).
The PAG influences descending pain modulation primarily through its reciprocal connections with the rostroventral medial medulla (RVM) (58). Excitation of PAG neurons also excites the activity of RVM neurons and is associated with inhibition of nocifensive spinal and craniofacial reflexes in the rat and cat (39,40,59). The RVM encompasses the nucleus raphe magnus, the nucleus reticularis gigantocellular-pars alpha, and the nucleus paragigantocelluraris lateralis (60). Stimulation of the RVM exerts predominantly inhibitory, but also facilitatory, effects, on nociceptive and non-nociceptive VBSNC neurons and orofacial sensory and motor behaviors (1,5,39,40,61). Reciprocal connections have been shown between the ventral part of the caudalis/interpolaristransition zone and the RVM, and direct projections also occur from the RVM to subnucleus caudalis. Through these connections, the activity of WDR and NS neurons can be either facilitated or inhibited, and perceptual, emotional, autonomic, and neuroendocrine responses to noxious stimuli can be totally or partially inhibited or amplified (i.e. facilitated) (1–5,39,40,62–68). Importantly, in cases where the balance of descending modulatory influences is disrupted in favor of facilitation, this process may promote and sustain central sensitization and pain chronicity (69). This has clinical significance, since evidence for enhanced descending facilitation has been reported in craniofacial pain states (70–72).
In addition to the PAG, the RVM also receives inputs from the thalamus, the parabrachial area and the noradrenergic locus coeruleus, and is considered to be the final common relay in descending modulatory pathways influencing pain, through its projections to the V subnucleus caudalis and to the caudalis/interpolaris transition zone, as well as the spinal dorsal horn. The RVM exerts a bidirectional pain modulatory effect, both inhibiting and facilitating pain. RVM “on-cells” have been characterized to increase their activity in response to noxious stimuli and prior to a nociceptive reflex, whereas “off-cells” cease firing immediately prior to the reflex (58,73,74). Opioids inhibit on-cells and cause excitation of off-cells, and the latter effect is considered “necessary and sufficient” to produce analgesia (58,74). The existence of on-cells and off-cells with descending projections to the V subnucleus caudalis and caudalis/interpolaris transitional zone, as well as the spinal dorsal horn, provides a neuronal substrate for positive and negative pain modulation from the PAG/RVM system (74). Moreover, as this system receives inputs from higher CNS sites, it also provides a mechanism whereby homeostatic or existential priorities may tone down or augment nociceptive inputs (74). Increased activity of V primary afferents, along with dysfunction in modulation from brainstem and diencephalic nuclei of V nociceptive afferent inputs, are likely to be essential components of several craniofacial pain disorders (47,75,76).
As noted above, an imbalance between the inhibitory and facilitatory descending pain modulatory systems may underlie pathological pain states including migraine (74,77–79). In rodent models of migraine-related pain, driving dural nociceptors with an inflammatory mediator cocktail in rats can result in activation of pain-facilitatory neurons within the RVM and with delayed, and generalized, facial allodynia, while inhibition of the RVM by microinjection of local anesthetics can reverse ongoing pain (80,81). In a preclinical model of medication overuse headache (MOH) it was demonstrated that morphine withdrawal may result in activation of pronociceptive neurons in the RVM and consequent increased activity of trigeminovascular neurons involved in headache pain (82). Dysfunction of endogenous pain-modulatory systems may also contribute to other chronic facial pain conditions, including TMD (4,70). A role for descending facilitation has also been reported in experimental V neuropathic pain (3,5,83). Direct corticotrigeminal projections from the insular cortex to the subnucelus caudalis have also been shown to facilitate the nerve injury-induced trigeminal neuropathic pain (84).
Diffuse noxious inhibitory controls of craniofacial pain and conditioned pain modulation
Brain-imaging studies have provided new insights into changes in brain function and circuits associated with chronic craniofacial pain (28). For example, there is growing consensus that migraine is a disorder of the brain (85). In patients suffering from trigeminal neuralgia, a significant reduction in grey matter volume of many structures associated with pain processing and perception, such as the thalamus, somatosensory cortex and insula, among others, has been observed with apparent correlation to disease duration (86–88). Likewise, in other V neuropathic pain states as well as TMD, there is evidence for structural changes in some of these CNS areas (89–91). These findings are consistent with the preclinical findings noted above of neuroplastic changes in animal models of chronic craniofacial pain. The clinical relevance of these neural adaptations has emerged from studies of the dynamic regulation of pain with either temporal summation or through the conditioned pain modulation (CPM) paradigm (92). Studies in animals have demonstrated the phenomenon whereby “pain inhibits pain” referred to as “diffuse noxious inhibitory controls” or DNIC (93). DNIC was first described in animals (93,94) but has now been demonstrated in humans as conditioned pain modulation (CPM) (92). It is believed that CPM may help to prevent further injury at the site of the most severe injury when multiple injuries are present so that the most important neural signals reach the brain. CPM occurs in the absence of distraction, and the analgesic component of distraction is additive with CPM (95).
Electrophysiological studies in rats have shown that the activity of neurons in the VBSNC can be strongly suppressed by application of an intensive pain stimulus outside their peripheral receptive field (61,66,96). Likewise, studies on healthy volunteers have shown that application of tonic painful conditioning stimuli in the craniofacial region may evoke DNIC-like mechanisms on segmental, as well as heterosegmental (i.e spinal) sites (97,98). DNIC mechanisms in animals involve an interaction between several brainstem structures, the dorsal reticular nucleus, RVMand PAG. The brain circuitry responsible for CPM in humans remains unknown, but recent findings support the involvement of brainstem structures, thus corroborating previous studies in this field (89).
Assessment of DNIC or CPM has important implications, as clinical studies have shown that many chronic or recurrent pain conditions such as TMD, as well as functional pain disorders such as migraine, chronic migraine and others may be due in part to a dysfunction of endogenous pain modulation and loss of the efficiency of CPM (99–103). Moreover, it has been suggested that the efficacy of the DNIC response in a rodent experimental neuropathic pain model was predictive of recovery, while in humans pre-operative assessment of CPM was used as a prospective predictor for chronic pain development after thoracotomy (104, 105) as well as for the efficacy of a reuptake blocker (i.e. duloxetine), which mimics descending inhibition in the treatment of pain (106).
Only a few studies have assessed the contribution of CPM impairment to chronic craniofacial pain conditions. Development of chronic tension-type headache and migraine has been associated with dysfunctional CPM (71,72). Patients with MOH also showed an altered CPM response compared to control subjects, which tended to improve after withdrawal of the drug that produced the MOH in the first place. The clinical evidence was corroborated in a preclinical model consisting of repeated application of inflammatory mediators to the rat dura mater (107). Persistent cephalic and extracephalic allodynia was observed along with increased FOS expression in the VBSNC and impairment of the DNIC response. Importantly, central sensitization indicated by the increased FOS expression, coupled with the loss of the DNIC response, was suggested as a mechanism that could elevate the risk for developing chronic migraine (107). In line with this observation, rats with sustained morphine-induced sensitization, a model of MOH, had a loss of the DNIC response in V subnucleus caudalis. Administration of lidocaine into the RVM, which abolishes descending facilitation, restored the DNIC response in the morphine-exposed rats, suggesting that the apparent loss of DNIC-related inhibition was due to enhanced facilitation (108). Another piece of evidence of CPM deficits associated with secondary headache was provided by a study in patients with traumatic brain injuries (TBI). In this study, patients who also developed chronic post-traumatic headache after TBI were demonstrated to have deficient CPM relative to TBI patients without headache (109). Reduced CPM has been also demonstrated in TMD patients (102,103,110). Taken together, these studies provide evidence that persistent craniofacial pain may be associated with dysfunction of endogenous pain modulatory systems with descending facilitation that may swamp descending inhibition to promote and sustain pain.
Clinical implications of central amplification for chronic craniofacial pain
Insights from experimental models and clinical observations have provided substantial evidence that mechanisms of amplification in the periphery as well as in the central nervous system can promote chronic pain conditions, including chronic craniofacial pain states. Reducing nociceptive afferent inputs into the CNS decreases central sensitization and chronic pain as shown by decreased receptive fields size and diminished referred pain and allodynia. Human brain imaging studies have also greatly advanced our understanding of brain circuits that modulate pain, and have revealed areas that have altered activity or connectivity patterns in V neuropathic pain and non-neuropathic pain states (e.g. TMD). They have also suggested possible sites that may be pain generators for migraine and for cluster headache (28,47,90,91,111). It was recently shown that hypothalamic activity in response to craniofacial stimulation (i.e., olfactory and visual) is altered during the 24 h prior to pain onset, and changes in the hypothalamo-brainstem connectivity may represent the driver of migraine attacks (112). Migraineurs may be identified by resting functional connectivity of certain brain areas, which are more evident in those with longer disease duration, suggesting a gradual reorganization of brain circuits with time (113). The volume of the anterior hypothalamus was shown to be enhanced in episodic and chronic cluster headache patients, suggesting its possible role as the cluster generator activating the trigemino-parasympathetic reflex (111,114). Deep-brain stimulation of the posterior hypothalamus has been shown to be clinically effective for drug-resistant chronic cluster headache, and has been explored for other complex craniofacial pain states (115,116).
It was noted above that a key mechanism of central sensitization in persistent craniofacial pain conditions is an imbalance of descending inhibition and facilitation. A shift to net descending facilitation appears to be essential in eliciting and sustaining chronic craniofacial pain that is related to injury, such as PTTN, or that is idiopathic (i.e. functional) in nature such as migraine (69). Indeed, changes in the PAG/RVM system or hypothalamus and their descending modulatory influences have been linked to several pain states including TMD, V neuropathic pain conditions, migraine and some types of headaches and have been also considered a predictor for the development of chronic craniofacial pain and other pain states (117,118). Understanding the underlying mechanisms that mediate adaptive changes in modulatory circuits is likely to be important for the development of novel pharmacological or non- pharmacological strategies to improve the management of craniofacial pain states.
Clinical implications
The craniofacial region is the site of some of the most common acute and chronic pain conditions that are often associated with special psychological and emotional meaning; management of these conditions can be very difficult. Chronic craniofacial pain conditions involve both peripheral and central amplification of nociceptive signals that may sustain pain. Repeated episodes of pain and sustained nociceptive drive may shift the balance of central modulation to favor facilitation, also promoting amplification and contributing to sustained chronic pain. Increased understanding of the mechanisms underlying trigeminal nociceptive transmission and its control can provide new insights for improved pain management.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support from the NIH-National Institute on Drug Abuse (DA034975 and DA041809, FP), National Institute of Craniofacial and Dental Research and Canadian Institutes of Health Research, BJS) is gratefully acknowledged.