
Abstract
Background
Blocking the pro-nociceptive action of CGRP is one of the most promising approaches for migraine prophylaxis. The aim of this study was to explore a role for CGRP as a neuroprotective agent for central and peripheral neurons.
Methods
The viability of isolated rat trigeminal, cortical and cerebellar neurons was tested by fluorescence vital assay. Engagement of Nrf2 target genes was analyzed by qPCR. The neuroprotective efficacy of CGRP in vivo was tested in mice using a permanent cerebral ischemia model.
Results
CGRP prevented apoptosis induced by the amino acid homocysteine in all three distinct neuronal populations. Using a set of specific kinase inhibitors, we show the role of multi-kinase signaling pathways involving PKA and CaMKII in neuronal survival. Forskolin triggered a very similar signaling cascade, suggesting that cAMP is the main upstream trigger for multi-kinase neuroprotection. The specific CGRP antagonist BIBN4096 reduced cellular viability, lending further support to the proposed neuroprotective function of CGRP. Importantly, CGRP was neuroprotective against permanent ischemia in mice.
Conclusion
Our data show an unexpected ‘positive’ role for the endogenous pro-nociceptive migraine mediator CGRP, suggesting more careful examination of migraine prophylaxis strategy based on CGRP antagonism although it should be noted that homocysteine induced apoptosis in primary neuronal cell culture might not necessarily reproduce all the features of cell loss in the living organism.
Introduction
Migraine is a common neurological disorder, with two main types distinguished by presence or absence of aura (1). Migraine aura is believed to be related to cortical spreading depression (CSD), a phenomenon whereby an initial robust excitation is followed by lasting neuronal depression (2,3). CSD-like events also develop in stroke patients, due to massive elevation of extracellular K+, neurotransmitter release and a strong influx of Na+ and Ca2+ into neurons (4). Familial hemiplegic migraine (FHM) affects both cortex and cerebellum (5), and CSD in this disorder is likely linked to excessively high levels of glutamate (6). NMDA receptors are primary contributors to CSD (3,7). Hyperactivation of NMDA receptors during CSD may result in neurotoxicity, especially if associated with hypoxia or hypoperfusion (8).
Accumulating evidence suggests that migraine with aura may be a risk factor for subclinical brain infarction (9,10). Some studies based on high-resolution imaging showed what could be considered as signs of neurodegeneration: Lesions and microinfarctions in migraine with aura (11). However, this issue is controversial as, according to other studies, the brains of migraineurs show no sign of damage (12) and migraine with aura is observed only in a fraction of migraineurs, whereas migraine without aura is a more common disorder.
Several studies have reported that migraine is associated with the release, both in the peripheral and central nervous system, of a 37-amino acid peptide calcitonin gene-related peptide (CGRP) stored in large size synaptic vesicles in various types of neurons (13, 14). CGRP receptors are Gs-coupled metabotropic receptors widely expressed in the central and peripheral nervous system (15,16). CGRP activates multiple signaling pathways including protein kinase A (PKA), protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase (CaMKII), leading to sensitization of receptors transducing pain-producing stimuli in trigeminal neurons (17,18). Apart from migraine, CGRP could be released during subarachnoid hemorrhage (19). High levels of CGRP could alleviate the damaging brain effect of ischemia, likely via its vasodilatory action (20). However, the direct neuroprotective effect of CGRP has never been tested. One suitable model to test this possibility is the neurotoxic action of homocysteine (HCY), high levels of which are associated with migraine and stroke and can lead to neuronal death through the activation of glutamate receptors (21).
Our study demonstrates that CGRP provides neuroprotection against HCY neurotoxicity for sensory, cortical and cerebellar neurons, and against ischemic brain damage in vivo. This remarkable neuroprotective effect of CGRP largely challenges further applications of anti-CGRP treatments.
Materials and methods
All experiments follow the Helsinki Declaration and guidelines set by the European Commission. The experiments were done under the licenses EKS-004-2014 and ESAVI-2015-000744 approved by the Committee for the Welfare of Laboratory Animals of the University of Eastern Finland and the Animal Experiment Committee in the State Provincial Office of Southern Finland. All animals were housed in cages with controlled temperature, humidity and 12-hour light-dark cycle. Food and water were provided ad libitum. ARRIVE guidelines were followed throughout the experiment. All efforts were made to minimize the number of animals used and their suffering.
Primary culture of rat trigeminal sensory neurons
Trigeminal ganglion (TG) cultures were prepared as described previously (21,22). Male Wistar rats at 10–12 postnatal days (P10–12) were sacrificed by CO2 inhalation. The trigeminal ganglions were isolated and enzymatically dissociated using 3% collagenase (Sigma) for 20 minutes. Cells were cultured in Ham's F-12 Nutrient Mix medium (Gibco) at 37℃, 5% CO2 for 48 hours prior to experimental treatment.
Primary culture of rat cortex
Primary cortical cultures were prepared as described earlier (23). P1–P2 Wistar rats were sacrificed after CO2 inhalation and cerebral cortices were isolated, enzymatically dissociated, and used for preparing primary neuronal cultures. Cells were cultivated in Neurobasal™ medium (Gibco) with B-27 supplement (Gibco) and used for experiments after 7–10 days in vitro (DIV).
Primary culture of rat cerebellar granule cells
Primary cerebellar granule cell culture was prepared as previously (24). Wistar P7 rats were sacrificed by CO2 inhalation and the cerebellums were isolated, enzymatically dissociated by trypsin and DNAse (Sigma), and used for preparing a primary culture. Cells were cultivated in Minimum Essential Medium (MEM, Gibco) with 10% fetal bovine serum (Gibco), together with 20 mM KCl (24) at 37℃, 5% CO2 for 7–10 DIV.
Experiment procedures
To test the viability, cultured cells were incubated for five or 24 hours in a normal medium or in a medium containing 500 µM homocysteine (HCY, D,L-form). Notably, only the L-form of HCY is biologically active, providing ∼50% of biologically active substance (21). HCY was also combined with rat α-CGRP (PolyPeptide group) or with the antagonist of CGRP receptors, BIBN4096 (Tocris Bioscience). CGRP was applied 20 minutes prior to HCY. The growth culture medium for the trigeminal culture contained 100 µM glycine (Ham's F-12 Nutrient Mix), whereas the cortex and cerebellar culture growth medium contained 75 µM glycine (Neurobasal™ medium). The MEM medium used for the cerebellar granule cell culture did not contain glycine. Therefore, in these experiments, we added 100 µM glycine to ensure the activation of NMDA receptors. To activate adenylate cyclase we used the specific activator forskolin (Sigma-Aldrich). The specific PKA inhibitor fragment 14–22, myristoylated trifluoroacetate, was used to block the cAMP signaling pathways, whereas chelerythrine was applied to inhibit PKC (all from Sigma-Aldrich) and KN93 (Tocris Bioscience) to inhibit CaMKII.
Quantitation of cell viability
Cell viability was measured by fluorescent viability assay (FVA) described earlier (23). In short, cells were treated with 0.001% acridine orange (Sigma) for 30 seconds in basic solution (152 mM NaCl, 2.5 mM KCl, 10 mM glucose, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, with the pH adjusted to 7.4 using NaOH). After complete washout of acridine orange, cells were exposed to 0.004% ethidium bromide (Sigma) for 30 seconds in basic solution, followed by dye washout. Fluorescence images were captured using a Leica TCS SP5 MP scanning confocal microscope (Leica Microsystems Inc.) or Olympus FV1000 confocal microscope (Olympus). For two-channel imaging, fluorescence was excited with a 488 nm laser and the emitted fluorescence was acquired at 500 to 560 nm (the green region of the spectrum for acridine orange) and above 600 nm (the red region of the spectrum for ethidium bromide). Single focal plane images from both channels were merged and analyzed with standard Leica LAS AF software (Leica Microsystems Inc.) and ImageJ software. On the resulting image, non-colocalized green and red pixels were attributed to live and necrotic neurons, respectively. Yellow-orange pixels with colocalized green and red fluorescence were attributed to the nuclei of apoptotic neurons (Figure 1a–c).
Neuroprotective effect of CGRP against homocysteine (HCY) induced neurotoxicity in rat trigeminal ganglion (TG) cells. An overlay of confocal images taken in the green and red spectra of TG cultures and correlation plots for the images showing live (green, L), necrotic (red, N) and apoptotic (orange, A) cells 24 hours in control (a), 24 hours in 500 μM HCY (b) and 24 hours in HCY together with 1 μM CGRP. (c) Histograms showing percentage of live cells in control and after 24 hours exposed to 1 μM CGRP, 500 μM HCY, 1 μM BIBN4096, as an antagonist of CGRP receptors, in different combinations. Mean ± SEM (n = 4–10), *p < 0.05, **p < 0.01, ***p < 0.0001 by Mann-Whitney U-test.
qPCR
Trigeminal cells from P10–12 rats were used one to two days after isolation. For experiments, the culture medium was replaced by a fresh one with or without 1 µM CGRP. Control and CGRP treated samples were collected at 90 minute time points. To test the effect of HCY on CGRP treated cells, 100 µM HCY was added to the medium 30 minutes after placing cells in a CGRP-containing or CGRP-free medium. Cells were collected 90 minutes later. Total RNA was extracted with TRI reagent® (Molecular Research Center, Inc). One microgram of the RNA was used for cDNA synthesis by Transcriptor First Strand cDNA synthesis kit (Roche). The relative expression levels were measured by qPCR, using the relative standard curve method (StepOnePlus Real-Time PCR Systems, Applied Biosystems) with specific rodent primers and probes (Roche) as follows: GclmF 5’tgactcacaatgacccaaaaga3’ – GclmR 5’ tttcacgatgaccgagtacct 3’ – probe #5; Nqo1F 5’ agcgcttgacactacgatcc 3’ – Nqo1R 5’ cgtgggccaatacaatcag 3’ – probe #50; Hmox1F 5’ gtcaagcacagggtgacaga 3’ – Hmox1R 5’ ctgcagctcctcaaacagc 3’ – probe #4; Nfe2l2F 5’ agcatgatggacttggaattg 3’ – Nfe2l2R 5’ cctccaaaggatgtcaatcaa 3’ – probe #3, Tubb-3 (beta tubulin3) F 5’ tatgtgcccagagccattct 3’ – Tubb-3R 5’ ttgttgccagcaccactct 3’ – probe #1. QPCR conditions: 95℃, 10 minutes; 40 × (95℃, 10 seconds; 60℃, 30 seconds).
Ischemia surgery and MRI imaging
The mice were randomized into treatment groups using GraphPad Prism QuickCalcs software-given random numbers. Mice (n = eight per group) were anesthetized with 5% isoflurane (in 30% oxygen – 70% nitrogen), and the surgical anesthesia was maintained with 2% isoflurane. The temperature of the mice was controlled by a homeothermic control system connected to a heating blanket connected to a rectal probe (Harvard apparatus; PanLab). The left middle cerebral artery (MCA) was permanently occluded as described for this model, called permanent middle cerebral artery occlusion (pMCAO, 25). Briefly, the temporal bone was first exposed and a 1 mm diameter hole was drilled to expose the MCA. The dura was carefully removed, after which the artery was lifted and occluded using a thermocoagulator (Aaron Medical Industries Inc, Clearwater). The occlusion of the MCA was confirmed by cutting the artery, after which the temporal muscle was replaced and the wound was sutured. The sham mice went through the same procedures except for the occlusion of the MCA. After the surgery, the mice were returned to their home cages to recover from anesthesia. CGRP (Sigma) was administered intravenously (iv) immediately after the surgery at a dose of 1 µg/mouse. Two mice died during the surgery due to disruption of the MCA. All other mice recovered well from the surgery and showed no adverse effects from the drug treatments.
The lesion size was imaged by magnetic resonance imaging (MRI) at 24 hours post stroke using a vertical 9.4 T Oxford NMR 400 magnet (Oxford Instrument PCL). Briefly, the mice were maintained under 2% isoflurane anesthesia. Twelve T2-weighted images were taken at 0.8 mm intervals (repetition time 3000 ms, echo time 40 seconds, matrix size 128 × 256, field of view 19.2 × 19.2 mm2). The lesion sizes were quantified from MRI images using home made software, Aedes, under the Matlab environment (MathWorks) by a researcher blind to the type of treatment. The lesion sizes were calculated using the published formula (26) and expressed as relative lesion volumes.
Statistical analysis
Imaging data were analyzed using the Mann-Whitney U-test with Bonferroni’s correction for multiple comparisons. The qPCR and in vivo stroke model data were analyzed using the two-tailed unpaired t-test. The results are expressed as the mean of data ± SEM (standard error of mean). The level of statistical significance was set to p < 0.05.
Results
CGRP prevents HCY induced toxicity in trigeminal, cortical and cerebellar neurons
To assess the neuroprotective effect of CGRP, trigeminal neurons were pre-treated with CGRP for 20 minutes prior to exposure to the neurotoxic amino acid HCY. Figure 1a shows the distribution of live, apoptotic and necrotic cells in control conditions. HCY applied for 24 hours significantly reduced the number of live cells to 44.2 % (Figure 1b, 1d). Notably, the toxic effect of HCY was prevented by 1 µM CGRP (Figure 1c, 1d). A selective antagonist of CGRP receptors BIBN4096 (1 µM) either alone or in combination with HCY, significantly reduced the amount of live cells (Figure 1d).
CSD is able to trigger intra-cortical CGRP release (3,27), which can potentially interfere with the survival of cortical neurons. In cortical neurons, HCY (500 µM) significantly decreased the proportion of live cells from a control level of 90.7% to 48.7% (Figure 2b, 2d). Pretreatment with CGRP rescued neurons from apoptosis evoked by HCY, since the number of live cells significantly increased by up to 72.3% (n = 5). This value differed significantly (p = 0.019) from the fraction of live cells exposed to HCY alone (Figure 2c, 2d). Whereas CGRP alone did not affect the cell viability (Figure 2d), the CGRP antagonist BIBN4096 (1 µM) significantly reduced the fraction of live cells to 56.9% (Figure 2d). CGRP failed to prevent the HCY-induced neurotoxicity in the presence of BIBN4096 (Figure 2d).
Neuroprotective effect of CGRP against homocysteine (HCY) induced neurotoxicity in rat cortical cells. An overlay of confocal images taken in green and red spectra of cortical cultures and correlation plots for the images indicating live (green, L), necrotic (red, N) and apoptotic (orange, A) cells 24 hours in control (a) 24 hours in 500 μM HCY (b) and 24 hours in HCY together with 1 μM CGRP. (c) Histograms showing percentage of live cells in control and after 24 hours exposed to 1 μM CGRP, 500 μM HCY, 1 μM BIBN4096, as antagonist of CGRP receptors, in different combinations. Mean ± SEM (n = 4–8), *p < 0.05 and **p < 0.01 by Mann-Whitney U-test.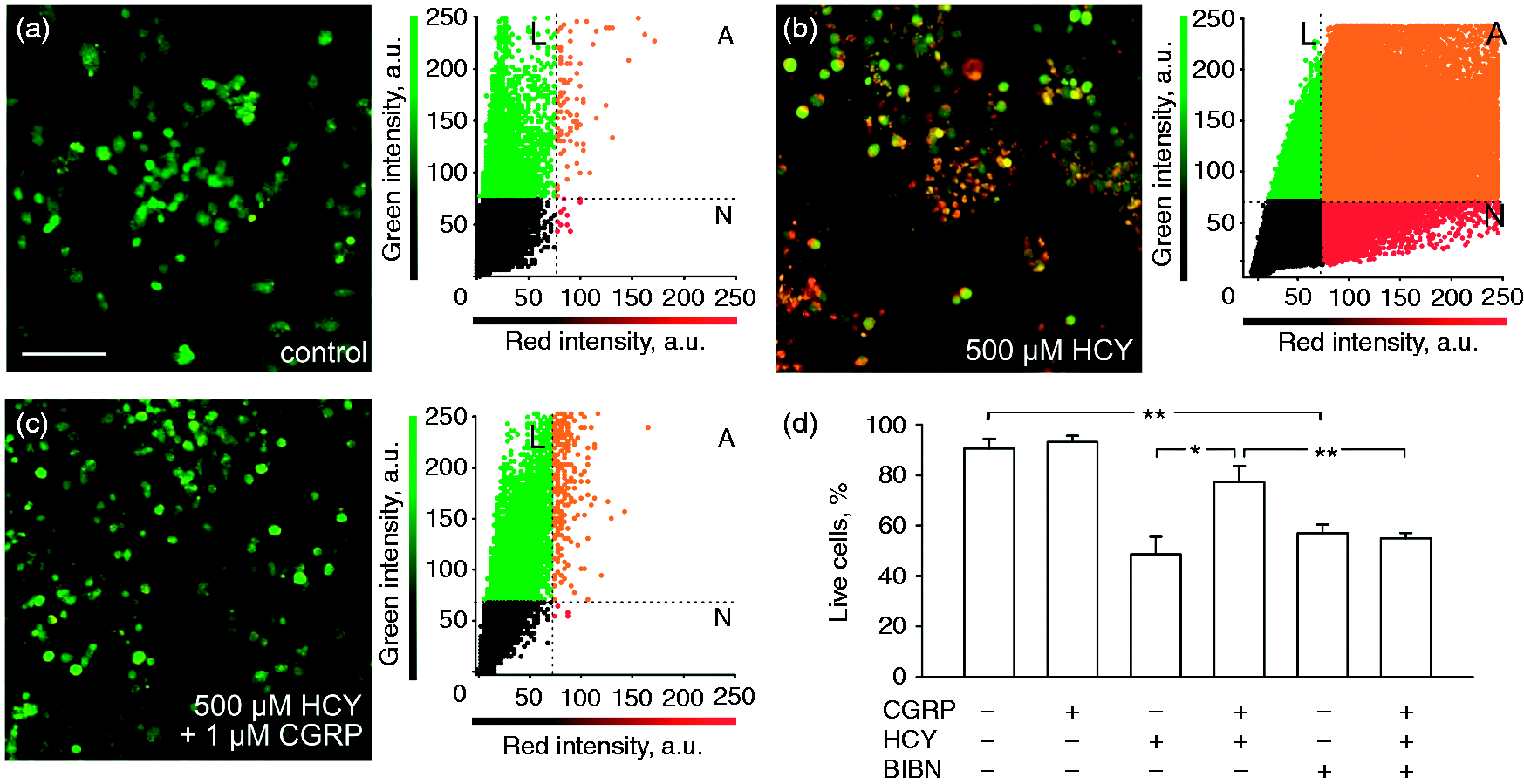
Accumulating evidence has suggested the involvement of the cerebellum in migraine pathology (5,28). Cerebellar cells widely express CGRP receptors (29), suggesting a signaling role for this neuropeptide in this brain region. HCY treatment (500 µM for 24 hours) significantly decreased the proportion of live cerebellar neurons (Figure 3b, 3d), but CGRP rescued these neurons from apoptosis (Figure 3c, 3d). Whereas CGRP alone did not affect survival (Figure 3d), 1 µM BIBN4096 significantly decreased the number of live cells (Figure 3d). CGRP failed to provide neuroprotection in the presence of BIBN4096 (Figure 3d).
Neuroprotective effect of CGRP against homocysteine (HCY) induced neurotoxicity in rat cerebellum cells. An overlay of confocal images taken in the green and red spectra of cerebellar cultures and correlation plots for the images indicating live (green, L), necrotic (red, N) and apoptotic (orange, A) cells 24 hours in control (a), 24 hours in 500 μM HCY (b) and 24 hours in HCY together with 1 μM CGRP. (c) Histograms showing percentage of live cells in control and after 24 hours exposed to 1 μM CGRP, 500 μM HCY, 1 μM BIBN4096, as antagonist of CGRP receptors, in different combinations. Mean ± SEM (n = 5–8), *p < 0.05, **p < 0.01, ***p < 0.0001 by Mann-Whitney U-test.
These observations suggest that endogenous CGRP is important for the survival of sensory, cortical and cerebellar neurons, and that CGRP receptors are essential for mediating neuroprotection by exogenous CGRP.
Signaling pathways involved in CGRP neuroprotection
Several kinases, such as PKA, PKC and CaMKII, are implicated in the pro-nociceptive action of CGRP (17). To investigate whether these kinases are important also for the CGRP-mediated neuroprotection, we assessed the effect of CGRP on HCY-induced neurotoxicity in the presence of PKA, PKC or CaMKII inhibitors. Whereas CGRP almost completely prevented the HCY-induced apoptosis of trigeminal neurons (Figure 4a), it failed to do so in the presence of the selective PKA inhibitor (0.6 µM; Figure 4a). The CaMKII blocker KN93 (3 µM) had a similar effect (Figure 4a). In contrast, the protective effect of CGRP was preserved in the presence of a broad spectrum PKC inhibitor, chelerythrine (1 µM; Figure 4a). Similar data were obtained in experiments with cortical neurons, in which the neuroprotective effect of CGRP was prevented by inhibition of PKA and CaMKII but not by PKC inhibition (Figure 4b). As in trigeminal neurons, the PKC inhibitor chelerythrine did not prevent the rescue effect of CGRP (Figure 4b). Interestingly, in cerebellar cells, all inhibitors (PKA, PKC and CaMKII) prevented the pro-survival effect of CGRP against HCY toxicity (Figure 4b).
Testing contribution of PKA, PKC and CaMKII in CGRP neuroprotection against homocysteine (HCY) neurotoxicity. Histograms showing percentage of live cells after 24 hours in control and after 24 hours’ exposure to 500 μM HCY alone or in combination with 1 μM CGRP and the peptide PKA inhibitor (0.6 μM), PKC inhibitor – chelerythrine (1 μM) or CaMKII inhibitor – KN93 (3 μM) in different combinations in (a) trigeminal ganglion (TG), (b) cortical (cortex) and (c) cerebellar cells (Crb) in vitro. Mean ± SEM (n = 4–10), *p < 0.05, **p < 0.01 by Mann-Whitney U-test.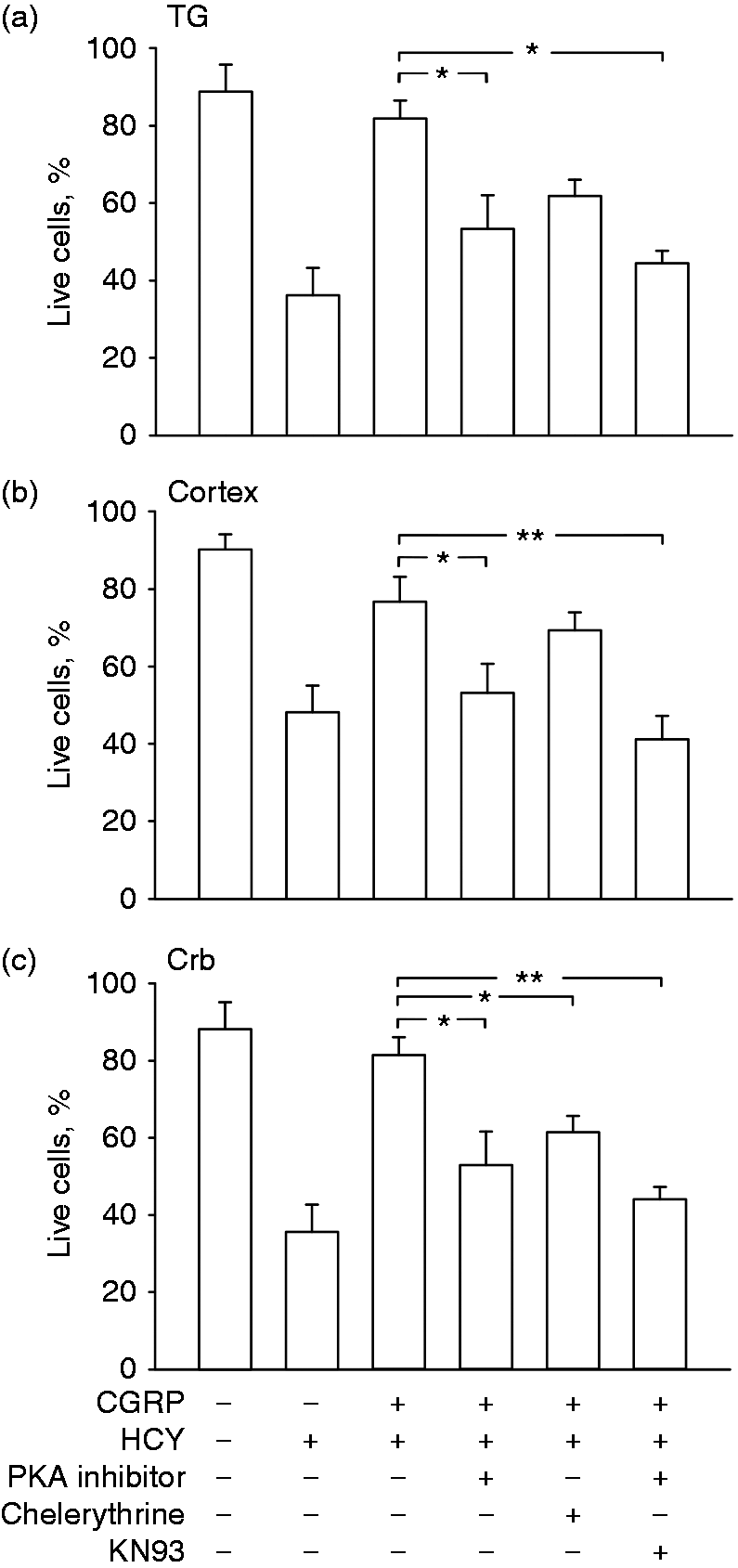
As CGRP is a strong inducer of cAMP (14), we next tested whether cAMP is indeed critical and sufficient for CGRP neuroprotection. To this end, we used forskolin as a classical activator of the adenylate cyclase/cAMP pathway. Figure 5 shows the noticeable neuroprotective efficacy of 1 µM forskolin in trigeminal, cortical and cerebellar neurons. Pre-application of the PKA inhibitor and CaMKII inhibitor KN93 prevented the forskolin-induced neuroprotection against HCY-induced toxicity, whilst the PKC inhibitor chelerythrine had no effect (Figure 5b–d). Thus, the direct activation of cAMP-dependent neuroprotective pathways by forskolin involves PKA and CaMKII, but not PKC activation.
Neuroprotective effect of forskolin (FK) against homocysteine (HCY) induced neurotoxicity. (a) An overlay of confocal images taken in the green and red spectra of trigeminal ganglion (TG) cultures and correlation plots for the images indicating live (green, L), necrotic (red, N) and apoptotic (orange, A) cells after 24 hour action of 500 μM HCY together with 1 μM forskolin (FK). Histograms showing the percentage of live cells in control and after 24 hours’ exposition to 500 μM HCY alone or in combination with 1 μM FK and with PKA inhibitor (0.6 μM), PKC inhibitor – chelerythrine (1 μM) or CaMKII inhibitor – KN93 (3 μM) in different combinations in (b) TG, (c) cortical (cortex) and (d) cerebellar cells (Crb) in vitro. Mean ± SEM (n = 4–10), *p < 0.05, **p < 0.01, ***p < 0.0001 by Mann-Whitney U-test.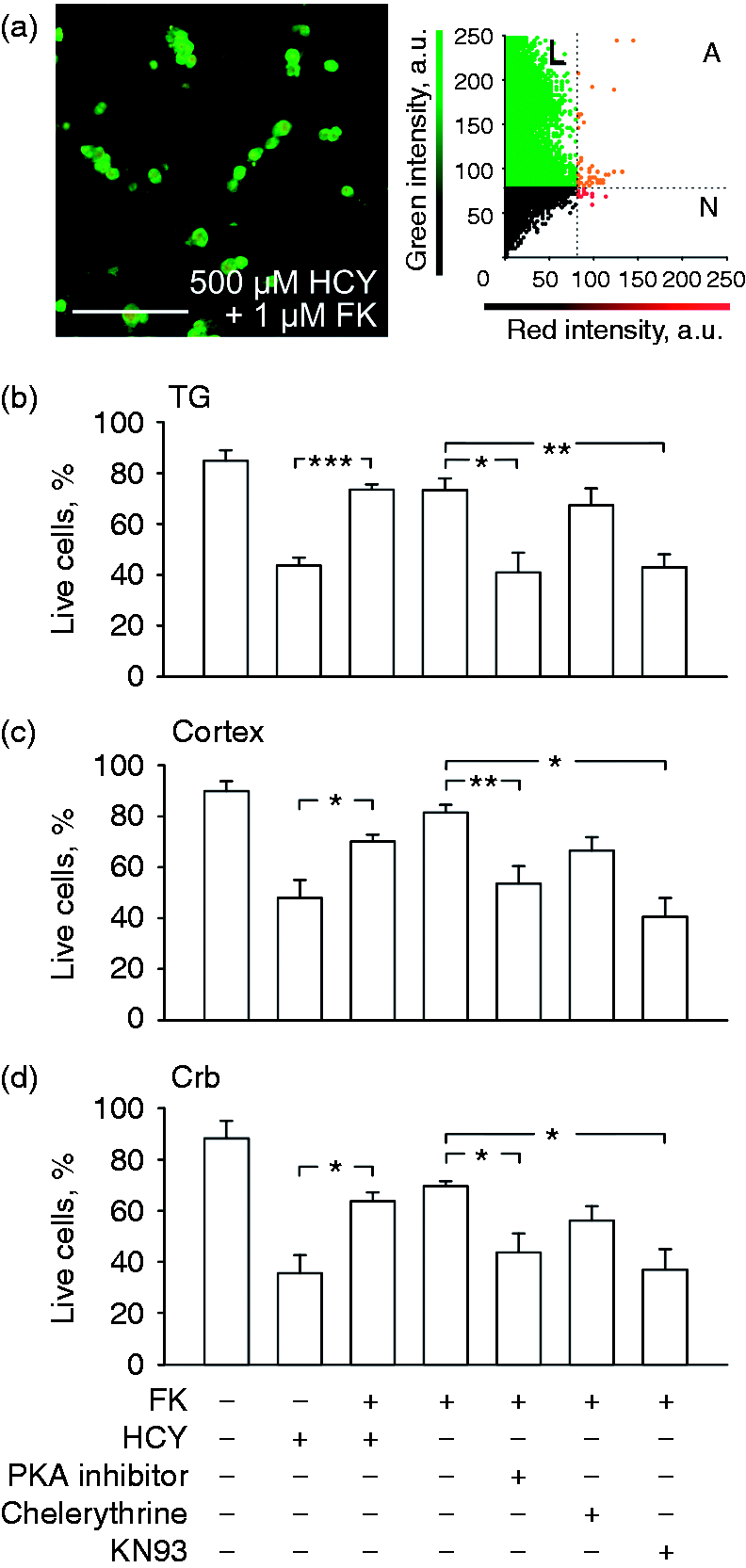
CGRP induced neuroprotection is not mediated through activation of Nrf2 pathway
As HCY can induce slowly developing oxidative stress in trigeminal and cortical cells (28), it is reasonable to suggest that the neuroprotection provided by CGRP could be mediated by the antioxidant system. The Nrf2-dependent transcriptional response is the key protective pathway that mediates neuroprotection in a number of neurodegenerative diseases (30). We therefore next tested whether HCY or CGRP affect the expression of Nrf2 and Nrf2 target genes, by using qPCR. Unexpectedly, CGRP failed to induce the expression of Nrf2 and heme oxygenase-1 (HMOX1), the modulatory subunit of γ-glutamylcysteine ligase (GLCM) and NAD(P)H:quinone oxidoreductase 1 (NQO1), the key Nrf2 target genes in trigeminal cells (Figure 6). Rather the opposite, while the level of Nrf2 mRNA was not significantly altered by HCY or CGRP, either alone or in combination (Figure 6а), the mRNA levels of NQO1, the key Nrf2 target gene that in part mediates the neuroprotective effects of Nrf2 in Alzheimer’s and Parkinson’s disease (31), was significantly decreased by co-treatment with HCY and CGRP (Figure 6b). Similarly, the expression level of HMOX1 was significantly reduced by CGRP and HCY (Figure 6c). Finally, GLCM expression was significantly decreased by HCY alone and by co-treatment with HCY and CGRP (Figure 6d). This data suggests that the Nrf2 pathway is not involved in CGRP neuroprotection.
Effect of CGRP, homocysteine and combined treatments on the antioxidant cell response in trigeminal cells. Histograms showing the expression level of Nrf2 (a), NQO1 (b), HMOX1 (c) and GLCM (d), in controls, 500 μM HCY, 1 μM CGRP or in combination of both agents. The expression was normalized to beta-tubulin and expressed as fold change relative to control. Mean ± SEM (n = 3), *p < 0.05, **p < 0.01 by two-tailed unpaired t-test.
CGRP is neuroprotective against ischemia induced cell death in vivo
To provide proof-of-concept of the neuroprotective efficacy of CGRP in vivo, CGRP was administered to mice subjected to pMCAO. At 24 hours post-surgery, quantification of the MRI images revealed a significant reduction in the lesion volume in CGRP treated mice compared to vehicle administered controls (Figure 7a). Figures 7b and 7c show typical examples of reduced lesion sizes in MRI images of vehicle (b) and CGRP-treated mice (c).
CGRP-induced significant neuroprotection in a mouse model of pMCAO. Quantification of the MRI images at 24 hours’ post stroke revealed a significant decrease in the lesion volume in CGRP treated mice compared to vehicle treated controls (a). Example MRI images of typical lesions of vehicle treated (b) and CGRP treated mouse (c), n = 6–8 per group, *p < 0.05 by two-tailed unpaired Student’s t-test.
Discussion
In this study, we show for the first time that the pro-nociceptive neuropeptide CGRP, which plays a key role in migraine pathology, may have important neuroprotective properties in peripheral and central neurons. Importantly, we show that CGRP is neuroprotective in a mouse model of pMCAO, providing a proof-of-concept of the in vivo neuroprotective capacity of CGRP. Thus, our findings are relevant not only to migraine but also to stroke, which is also often accompanied by CSD-like events in peri-infarct regions (8).
First, we assessed the neuroprotective effect of CGRP in a pertinent in vitro model of HCY-induced toxicity (21). Elevated levels of HCY are found in the plasma of migraineurs with aura (32), and carriers with the C677T genotype of the methylenetetrahydrofolate reductase (MTHFR) gene have increased levels of HCY (33) and an increased risk of migraine, especially migraine with aura (34).
CGRP showed prominent neuroprotective effects in all neurons tested, including peripheral sensory and central cortical and cerebellar neurons. Both cortical (3,16) and cerebellar (29) neurons express CGRP and CGRP receptors. Interestingly, the level of endogenous HCY in rats is several times higher in the cerebellum than in the other parts of the brain (35), making it potentially highly vulnerable to NMDA-mediated excitotoxicity (36).
CGRP is a 37-amino acid peptide stored in large size synaptic vesicles in various types of neurons (14). It is released in a Ca2+-dependent manner following activation of various membrane receptors (37). CGRP effects are mediated via activation of AC/cAMP/PKA, via the Gs-protein in different cell types (14). It has been shown previously that neuroprotection by PACAP can also be provided by activation of Gs-coupled receptors (38). Consistent with a key role for cAMP in neuroprotection, we found that the inducer of AC, forskolin, mimicked the neuroprotective effect of CGRP. Notably, both forskolin and CGRP provided this effect in a PKA- and CaMKII-dependent manner. These findings are in agreement with data from others showing that cAMP signaling, apart from PKA, also involves other kinases (38). Remarkably, PKA, PKC and CaMKII were also involved in the pro-nociceptive effect of CGRP, leading to the sensitization of pain transducing receptors in trigeminal neurons (17,18). Thus, CGRP induced neuroprotection was not mediated through the Nrf2 signaling pathway, but required the concerted activation of PKA, CaMKII.
The finding of the neuroprotective role of CGRP extends our understanding of migraine pathology, suggesting that the release of CGRP during migraine attack has an important biological role directed to neuronal survival. This view is consistent with the recent observation that the brains of migraineurs with aura do not suffer from widespread apoptosis after a potentially damaging event like CSD, which is usually associated with aura (39). Notably, CSD-induced preconditioning is neuroprotective in ischemic stroke (40) and CGRP has been proposed to play a role in cardiac preconditioning (41–43).
One important finding was that the specific CGRP antagonist BIBN4096 reduced neuronal survival in cortical and cerebellar neurons, indicating a neuroprotective role for endogenous CGRP. Notably, BIBN4096, and several other pharmacological CGRP antagonists, have been found to be effective for treatment of migraine (44), but their clinical applications were limited by unexpected liver toxicity and currently none of them is allowed for clinical use (45). Given the neuroprotective effect of CGRP, our study provides a rationale for the potential toxic effects arising from widespread CGRP antagonism. CGRP has been suggested as beneficial also in subarachnoid hemorrhage by counteracting vasoconstriction (19,46). Blocking CGRP is thus likely to have cardiovascular risks, as reviewed in detail in (47). Taken together, our data indicate that CGRP signaling could be considered to be a fundamental neuroprotective mechanism for central and peripheral neurons.
Conclusion
Here we show for the first time a novel neuroprotective role for CGRP in multiple neuronal populations against neurotoxicity induced by the amino acid HCY, which is implicated in migraine and several neurodegenerative disorders. This protective effect is mediated mainly through activation of the PKA and CaMKII pathways. Our data highlight the importance of CGRP signaling on neuronal survival, and suggest that migraine treatment strategies concentrating on blockage of CGRP should take into account the neuroprotective effect of this neuropeptide, although it should be noted that apoptosis in primary neuronal cell cultures might not necessarily reproduce all the features of cell loss in the living organism.
Article highlights
A novel role of CGRP as an endogenous neuroprotective agent for central and peripheral neurons is proposed. CGRP provides neuroprotection by cAMP activation involving PKA and CaMKII signaling. CGRP antagonism by BIBN4096 reduces the viability of isolated sensory and central neurons.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the Finnish Academy grant 277442 to RG, Russian Foundation for Basic Research grant 16-04-00653 to PAA and Russian Science Foundation grant 16-15-10192 to SMA. TM is supported by the Emil Aaltonen Foundation and the Academy of Finland.