
Abstract
Background
Tolosa-Hunt syndrome (THS) is characterized by unilateral orbital pain, ipsilateral oculomotor paresis and a prompt response to treatment with corticosteroids. Several reports have demonstrated that the clinical features of THS are not specific to one causal aetiology and can lead to misdiagnosis.
Case report
We report the case of a patient diagnosed with THS after an episode of unilateral orbital pain and diplopia with demonstration of granulomatous inflammation of both cavernous sinus on cerebral magnetic resonance imaging and an immediate response to treatment with corticosteroids. Progression of the disease over the following years, accompanied by increasing signs of inflammation on cerebral magnetic resonance imaging and cerebrospinal fluid pleocytosis, led to further diagnostic tests. Genetic analyses revealed a heterozygote low-penetrance mutation (Q703K) of the cryopyrin/NLRP3 gene compatible with a cryopyrin-associated periodic fever syndrome.
Discussion
This case report demonstrates that THS can be a central nervous system manifestation of cryopyrin-associated periodic fever syndrome, which therefore represents a differential diagnosis of THS, even in elderly patients.
Keywords
Background
Diagnostic criteria of Tolosa-Hunt syndrome (7).

A review of the published literature on THS from 1988 to 2002 demonstrated that only 21% of 208 reported cases were classified as THS based on these criteria (4). Possible differential diagnoses include recurrent cranial neuropathy, lymphoma, idiopathic hypertrophic cranial pachymeningitis, vasculitis, infectious disease, a tumour or sarcoidosis. This illustrates that the clinical features of THS are not specific, do not guarantee a correct diagnosis and may even can create a dangerous sense of false security, as illustrated in the following case report.
Case report
In June 2009, a 63-year-old Croatian man developed, within days, unilateral, left-sided, unbearable orbital pain accompanied by diplopia. The double vision ceased after one week, but the pain persisted and led to a first neurological consultation. The differential diagnosis of a trigemino-autonome headache was considered; however, a therapeutic trial with high-dose oxygen, sumatriptan and indomethacin gave no pain relief. Lumbar puncture results showed a normal cerebrospinal fluid (CSF) cell count (3 cells/µl) and normal CSF protein, but positive oligoclonal bands. C reactive protein (CRP) and the erythrocyte sedimentation rate (ESR) were both increased, although there was no other evidence of rheumatological disease. Cranial MRI demonstrated a granulomatous inflammation of both cavernous sinus, which was more pronounced on the left side, and a granulomatous inflammation of the anterior clinoid process and the cerebellopontine angle. The diagnosis of THS was made and treatment with corticosteroids (glucocorticoid, prednisolone 100 mg/day) was started. This immediately relieved the pain, supporting the diagnosis of THS.
The sinistral orbital pain reoccurred intermittently without diplopia when the steroid treatment was reduced and always showed an immediate response to the patient’s self-medication with a single dose of 20 mg prednisolone. However, the symptoms worsened with almost daily headache and nightly pain attacks from December 2014 and therefore cranial MRI was repeated and showed increasing signs of inflammation with a contrast-enhancing mass in both cavernous sinus, the left side of the basal dura and the left tentorium (Figure 1A), and an oedema in the left temporal lobe (Figure 1B). Lumbar puncture showed a CSF pleocytosis (24 cells/µl; 57% lymphocytes, 29% neutrophils and 10% monocytes) and increased CSF protein (110 mg/dl). CRP and ESR were repeatedly elevated.
(a) Axial T1-weighted image after the intravenous administration of a gadolinium-based contrast medium showing pronounced tentorial thickening and meningoencephalitic inflammation with granulomatosis-like enhancement. (b) Axial T2-weighted sequence demonstrating temporo-basal oedema.
This disease progression led to admission to our department. The patient was treated on admission with non-steroidal anti-inflammatory drugs, but no glucocorticoid. A neurological examination still showed no neurological deficit, but the patient reported daily, left-sided retro-orbital headache. A repeated lumbar puncture showed a normal CSF cell count (4 cells/µl) and normal CSF glucose, but increased CSF protein (86 mg/dl) and intrathecal IgG, IgA and IgM synthesis with positive oligoclonal bands. Angiotensin-converting enzyme was normal in the serum samples and CSF, s-IL2 receptor was mildly increased to 842 kU/l (standard value 158–623 kU/l) in serum samples, but a positron emission tomography-computed tomography scan with 18F-fluorodeoxyglucose showed no evidence of an organic manifestation of sarcoidosis. Antibody indices of HSV-, VZV-, CMV-, measles-, rubella- and lues-IgG, and borrelia-IgM and -IgG were normal; microbiological analysis showed no sign of central nervous system (CNS) tuberculosis with negative polymerase chain reaction, culture and microscopy. Inflammatory markers in serum, including CRP, ESR and serum amyloid A, were normal.
The constellation of recurrent episodes with increased inflammatory markers, unusual but distinct MRI features with meningeal contrast enhancement, the immediate response to treatment with glucocorticoids and temporary CSF pleocytosis prompted us to consider a CNS manifestation of an autoinflammatory syndrome, specifically a cryopyrin-associated periodic fever syndrome (CAPS). Genetic testing for the most prevalent syndromes (including familial Mediterranean fever syndrome, tumour necrosis factor receptor I associated periodic syndrome and CAPS) were performed and showed a heterozygote carrier status of a glutamine703 (CAG) > lysine (AAG)/p.Gln703Lys/Q703K substitution encoded by exon 3 of the NLRP3 gene, confirming the diagnosis of CAPS. The patient’s past medical history revealed (apart from headache) no other typical systemic manifestation of CAPS, such as myalgia, arthralgia, recurrent fever episodes, urticarial rash or gastrointestinal symptoms. An eye examination showed no ocular manifestation such as papillitis, conjunctivitis or uveitis, but audiometry revealed a bilateral high-frequency dip, indicating sensorineural hearing loss.
Finally, the diagnosis of CAPS-associated CNS inflammation manifesting as THS was made and high-dose corticosteroid treatment (a total of 5 g of methylprednisolone, followed by a slow oral reduction) was administered. This immediately stopped the headache. Six weeks later the patient was still asymptomatic and a follow-up MRI scan showed improvement with diminished contrast enhancement and regression of the oedema (Figure 2). As a result of recently occurring headache attacks with repeated glucocorticoid treatment and evidence for sensorineural hearing loss, anti-interleukin-1 treatment is currently being evaluated.
(a) Six weeks after administration of glucocorticoids, repeated magnetic resonance imaging showed improvement of the central nervous system inflammation with reduced contrast enhancement on axial T1-weighted image after the intravenous administration of a gadolinium-based contrast medium with (b) remission of the oedema in the left temporal lobe on an axial T2-weighted image.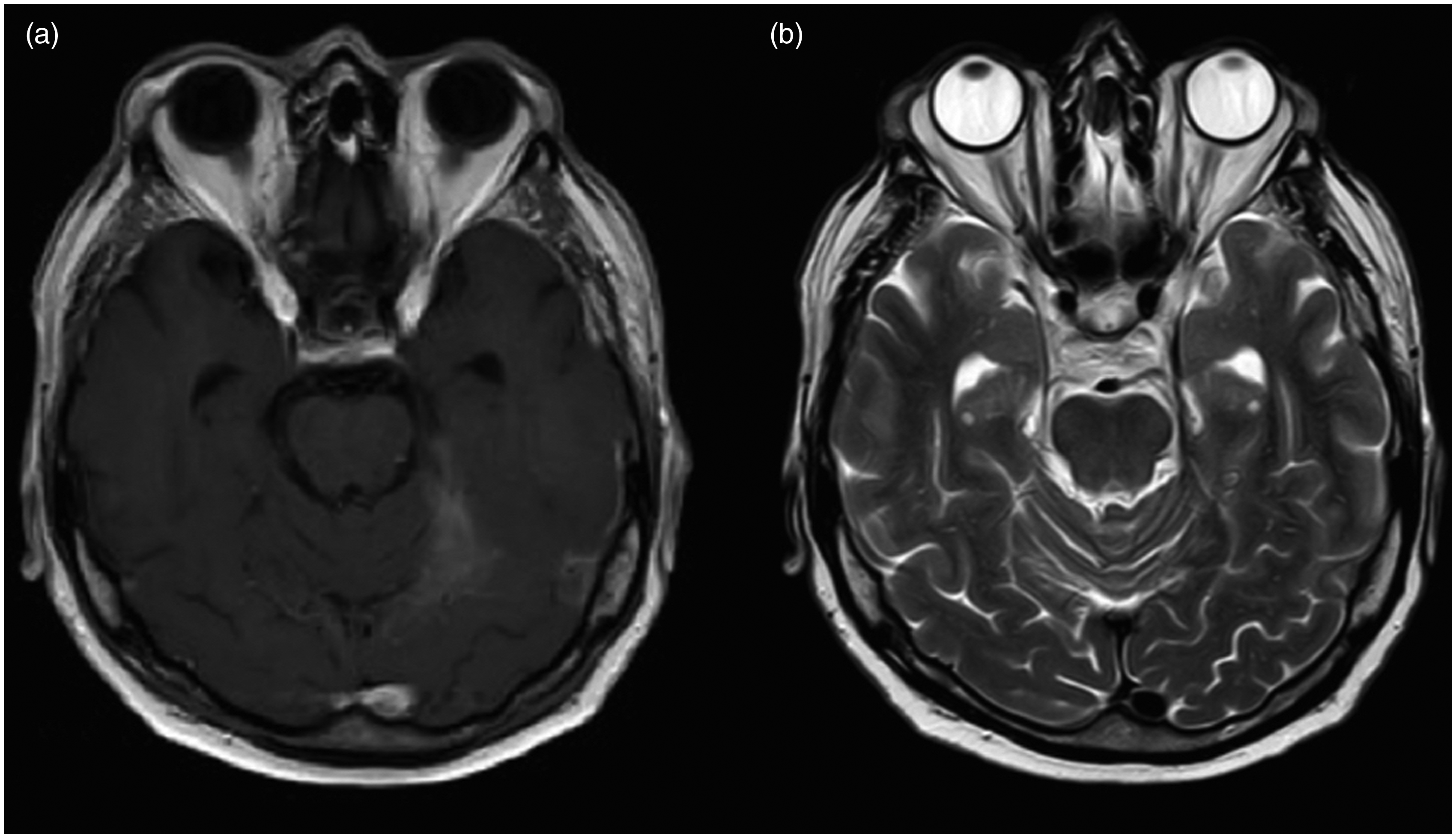
Discussion
This case report reflects the difficulties in establishing the diagnosis of THS. At the start of treatment, the diagnosis of THS was made on the basis of: (a) the initial clinical presentation with unilateral orbital pain and double vision; (b) the demonstration of a granulomatous inflammation of both cavernous sinus (more pronounced on the left side) on MRI; and (c) the immediate resolution of pain after treatment with corticosteroids. The patient later developed recurrent episodes of unilateral orbital headache without diplopia, which promptly disappeared on treatment with corticosteroids. Apart from increased inflammatory markers, no other associated symptom or clinical finding was present. Thus the patient completely fulfilled the criteria for THS. Progression of CNS inflammation years later with distinct MRI features, including an additional contrast-enhancing mass in the left side of the basal dura and the left tentorium and an oedema in the left temporal lobe, prompted further diagnostic workup. Molecular genetic analysis showed a Q703K mutation encoded by exon 3 of the NLRP3 gene, which is associated with CAPS.
CAPS are a group of rare, systemic monogenetic inherited autoinflammatory diseases. They encompass familial cold-induced autoinflammatory syndrome, Muckle–Wells syndrome and neonatal-onset multisystem inflammatory disease/chronic infantile neurological, cutaneous and articular syndrome (8,9). They are all caused by gain-of-function mutations in exons 3, 4 or 6 of the NLRP3 gene (formerly named CIAS1) encoding the protein cryopyrin, a component of the NLRP3 inflammasome. Overproduction of the proinflammatory cytokine interleukin-1 beta leads to multisystemic inflammation affecting the skin, eye, joints, bones and CNS (8). Patients may present with variable degrees of recurrent unprovoked episodes of fever, abdominal pain, myalgia, arthralgia and urticarial rash, as well as conjunctivitis or uveitis (10). CNS manifestations may also occur and include recurrent aseptic meningitis, increased intracranial pressure, headache syndromes, papilloedema and seizures, as well as vision and sensorineural hearing loss. In one report it was shown that patients with classical NLRP3 mutations and CNS manifestations mainly suffered from headache syndromes and papilloedema (9).

+++: severe; ++: moderate; +: mild; −:none; CN: cranial nerve; CNS: central nervous system.
Apart from CNS manifestations and recurrent elevation of inflammatory markers, we found little evidence for a systemic manifestation of CAPS. Atypical presentation and manifestation later in life can make neurological manifestations in patients carrying low-penetrance cryopyrin/NLRP3 mutations a difficult diagnostic challenge. Furthermore, neurologists are not usually familiar with CAPS. This constellation may lead to misdiagnosis and there may be a large number of unreported or misdiagnosed patients. Our case report illustrates that CAPS should be considered in patients with THS and recurrent episodes of aseptic meningoencephalitis. Both CAPS and THS show episodic exacerbations and respond rapidly to treatment with glucocorticoids. Careful evaluation of the patient’s medical history for additional unexplained recurrent symptoms, such as myalgias/arthralgias, urticarial rash, uveitis/conjunctivitis and repeated elevation of inflammatory markers, is necessary.
At the time of the initial presentation, the patient fulfilled the ICHD-II criteria for THS. However, the later ICHD-3 beta criteria for THS strengthened the diagnostic role of MRI and attenuated the role of the therapeutic response to treatment with glucocorticoids (13). Retrospectively, the patient’s first symptoms would still fulfil the current ICHD-3 beta criteria, but, later on, the clinical course changed and revealed the misdiagnosis. Therefore we recommend considering a period of follow-up examination before diagnosing THS, including repeated MRI and CSF diagnostics and repeated examinations for possible systemic manifestations of autoinflammatory disease.
Based on our experience with the reported patient, it might even be the case that an unknown number of patients diagnosed with THS and fulfilling all the IHS criteria may have CAPS as the underlying cause. Consequently, we recommend molecular genetic testing in patients with THS and additional symptoms or signs suggestive of CAPS, especially because additional treatment options other than glucocorticoids are available for patients with CAPS.
Clinical implications
In patients with Tolosa-Hunt syndrome, careful evaluation of their medical history with additional unexplained recurrent symptoms, such as myalgias/arthralgias, urticarial rash, uveitis/conjunctivitis and repeated elevation of inflammatory markers, is necessary. Molecular genetic testing should be considered in patients with THS and additional unexplained symptoms, even in older patients, because alternative therapeutic options (e.g. anti-interleukin-1 treatment) are available for cryopyrin-associated periodic fever syndrome.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.