
Abstract
Introduction
Familial hemiplegic migraine (FHM) is a rare autosomal dominant subtype of migraine with aura. The FHM3 subtype is caused by mutations in SCN1A, which is also the most frequent epilepsy gene encoding the voltage-gated Na+ channel NaV1.1. The aim of this study was to explore the clinical, genetic and pathogenetic features of a pure FHM3 family.
Methods
A three-generation family was enrolled in this study for genetic testing and assessment of clinical features. Whole cell patch-clamp was performed to determine the functions of identified mutant NaV1.1 channels, which were transiently expressed in human tsA201 cells together with β1 and β2 subunits.
Results and conclusions
We identified a novel SCN1A (p.Leu1624Pro) mutation in a pure FHM family with notably early-onset attacks at mean age of 7. L1624P locates in S3 of domain IV, the same domain as two of four known pure FHM3 mutations. Compared to WT channels, L1624P displayed an increased threshold-near persistent current in addition to other gain-of-function features such as: a slowing of fast inactivation, a positive shift in steady-state inactivation, a faster recovery and higher channel availability during repetitive stimulation. Similar to the known FHM3 mutations, this novel mutation predicts hyperexcitability of GABAergic inhibitory neurons.
Introduction
Familial hemiplegic migraine (FHM) is a rare autosomal dominant migraine subtype associated with aura including reversible hemiparesis (1,2). FHM has a remarkable phenotypic variability among patients even within the same family. Clinical onset is usually during youth. So far three causative genes have been identified, all encoding ion transport proteins: CACNA1A (FHM1) (3), ATP1A2 (FHM2) (4) and SCN1A (FHM3) (5). Recently PRRT2 has also been found to be associated with hemiplegic migraine (6). Mutations in these four genes are postulated to influence the initiation and propagation of cortical spreading depression (CSD), as evidenced by data from gene-targeted mouse models. CSD is a short-lasting propagating wave of cellular depolarization followed by a long-lasting suppression of the neuronal activity that is believed to be the basis for the migraine aura (7). The likelihood of CSD is increased by hyperexcitability of the neuronal network (8,9). In FHM1, CaV2.1 mutations induce a gain-of-function resulting in enhanced glutamate release and facilitated CSD. In FHM2, mutations of the sodium potassium ATPase cause a loss of function, leading to accumulation of extracellular K+ and the development of CSD (8,9). In FHM3, NaV1.1 mutations predict increased firing and a hyperexcitability of GABAergic neurons. This may also lead to an increase in extracellular K+ and thereby trigger CSD (10,11).
SCN1A, the gene encoding NaV1.1, is the most relevant gene in epilepsy with hundreds of mutations linked to a spectrum of epilepsy syndromes of varying severity (12). Previous studies of these epileptic mutations revealed mainly loss-of-function effects, particularly in mouse models. These studies predict network hyperexcitability due to specifically reduced action potential firing in inhibitory GABAergic interneurons, in which NaV1.1 is predominantly expressed (13–15). In vitro studies, also mainly loss-of-function effects were found, whereas a few studies also reported partial gain-of-function effects (16). Until now, only eight SCN1A missense mutations have been described to cause FHM3. Of those, some mutations (Q1489K, L1649Q, I1498M and F1661L) were identified in pure FHM families (5,17,18), whereas others were described to be linked to FHM associated with either epileptic seizures (L263Q, T1174S and Q1489H) or elicited repetitive daily blindness (Q1489H and F1499L) (19–21). Functional investigation of FHM3-causing SCN1A mutations revealed that most had gain-of-function effects such as increased non-inactivated depolarization-induced persistent sodium currents, delayed entry into inactivation, accelerated recovery from fast inactivation and rescue of folding defects by incubation at lower temperature or co-expression of interacting proteins (5,10,11,17,22). In contrast, the FHM3 mutation T1174S in NaV1.1, which incidentally caused a mixed FHM3 and epilepsy phenotype, showed both gain and loss of function (21).
In the present study, we identify a novel NaV1.1 L1624P mutation in a pure FHM family with early age at clinical onset. Electrophysiological studies show an overall gain of function of this mutation in agreement with the previously assumed pathomechanism of FHM3.
Materials and methods
Genetics
A three-generation family with five FHM patients was enrolled in this study, and the clinical features were analyzed. Their blood samples were collected for mutation screening after informed consent was obtained. All procedures were approved by the Ethics Committee of Ulm University and were in accordance with the Declaration of Helsinki. The mutation screening was performed by direct sequencing of all coding exons and exon-intron junctions of CACNA1A, ATP1A2 and SCN1A genes. SCN1A mutation nomenclature is based on reference transcript NM_001165963.1.
Functional studies
The shorter isoform (−11 amino acids) of the human NaV1.1 α subunit subcloned to the pCDM8 vector was used for site-directed mutagenesis of L1613P (equivalent mutation L1624P). SCN1A was transiently transfected in human tsA201 cells together with the auxiliary subunits β1 and β2 coupled to green fluorescent protein (GFP) and CD8 antigen, respectively. Whole-cell patch clamp recordings were performed 24 hours after transfection. Only cells expressing both reporters and producing voltage-dependent inward sodium currents as previously reported were used and analyzed (23). The pipette resistance was around 1.5 MΩ after filling with internal solution containing (in mM): NaF 10, CsF 110, CsCl 20, ethylene glycol tetraacetic acid (EGTA) 2, HEPES 10, NaATP 2 and NaGTP 0.2. The external solution contained (in mM) NaCl 145, KCl 4, CaCl2 1.8, MgCl2 1 and HEPES 10. pH was adjusted to 7.3 and osmolarity to ∼300 mOsm. Before data acquisition, cells were allowed to stabilize for 10 min after establishment of the whole-cell configuration. Sodium currents were recorded at room temperature (21–23℃) after partial series resistance compensation (∼85%) using an Axopatch 200B amplifier (Axon Instruments). The voltage errors were <5 mV. Data were filtered at 10 kHz and sampled at 50 kHz. Only amplitudes of peak sodium currents between 1.3 to 14 nA were used for analysis. All recordings were performed without online leak subtraction. The linear passive currents at various potentials were calculated from the value at the holding potential of −120 mV, and these currents were subtracted from the current recordings.
Data analysis
The steady-state activation was determined by fitting the peak current-voltage relation with the equation

Time constants of fast inactivation were obtained by fitting a double exponential function to the current decay

Steady-state inactivation curves were evaluated by fitting the data to the equation
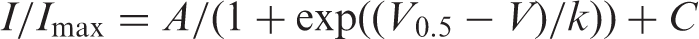
The recovery from inactivation was analyzed by fitting the data with a double exponential function:

Entry into slow inactivation was fitted by a single decay function

Data were analyzed by a combination of pClamp (Axon Instruments), Excel (Microsoft), SPSS (IBM) and ORIGIN (MicroCal software). Data are presented as mean ± standard error of the mean (SEM). Student’s t-tests were applied for statistical evaluation with significance levels set to *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
Phenotype and genotype
The three-generation German family contained five patients with migraine attacks fulfilling International Headache Society (IHS) criteria for FHM (Figure 1(a)). All patients, except the deceased grandfather of the index patient, underwent a detailed semi-structured individual personal interview or telephone interview, putting particular emphasis on the occurrence and severity of motor aura symptoms. Detailed clinical characteristics are depicted in Table 1.
FHM family with a novel SCN1A-L1624P mutation. Clinical features of the FHM3 family. Deceased. ID: identification; FHM3: familial hemiplegic migraine 3; NK: not known; NA: not applicable; H: hemiplegic; S: sensory disturbance; V: visual disturbance; A: aphasia; MO: migraine without aura; HP > HA: hemiplegic attack precedes headache; R: right side; L: left side.

All four tested affected family members (I.2, II.2, II.3, III.1) harbor a heterozygous T to C substitution at nucleotide 4871, corresponding to an L1624P mutation in segment S3 of domain IV of NaV1.1 (Figure 1(b)). This variant has not been reported in >120,000 alleles included in the ExAC browser. No mutations were detected in CACNA1A and ATP1A2 genes. III.2 did not attend the genetic test.
Electrophysiology
Representative whole-cell currents recorded from tsA201 cells expressing wild-type (WT) or mutant channels were elicited by various test potentials from a holding potential of −120 mV (Figure 2(a)). Peak current density of the mutant channels was similar to that of the WT channels (L1624P: −391.11 ± 56.46 pA/pF vs WT: −401.88 ± 47.93 pA/pF, Figure 2(a)). Likewise, voltage dependence of activation revealed no significant difference between WT (V0.5 = −23.93 ± 1.18 mV) and mutant L1624P channels (V0.5 = −25.36 ± 1.04 mV). The time course of fast inactivation was fitted to a second-order exponential function with currents being elicited by test pulses between −40 and 20 mV. There was a significant slowing of mutant channels, especially of the fast component (Figure 2(b)); i.e. at a test potential of −10 mV eliciting the maximum peak current, τfast of the mutant L1624P (1.21 ± 0.09 ms) was around 2.4 times slower than that of WT channels (0.52 ± 0.02 ms). This finding suggests a destabilization of the inactivated state of NaV1.1 channels. In agreement with this hypothesis, mutant channels showed a significant depolarizing shift of the steady-state fast inactivation curve (V0.5 = −49.10 ± 1.18 mV) compared with WT NaV1.1 channels (V0.5 = −58.29 ± 1.17 mV) (Table 2).
Functional characteristics of WT (filled symbols) and mutant L1624P (open symbols) channels expressed in tsA201 cells. Gating parameters of WT and mutant channels. Significance levels were set to *p<0.05, ap < 0.001. WT: wild-type.

To investigate the recovery from inactivation states, cells were depolarized to −10 mV for 100 ms to inactivate Na+ channels and then repolarized to −120 mV recovery potential for increasing duration (Figure 2(d), inset). Its time course was well fit to a second-order exponential function, yielding two time constants, τfast and τslow (Figure 2(d)). The faster time constant τfast was significantly decreased for mutant compared with WT channels, whereas the slower component τslow was similar between WT and mutant channels. For the fast component, the mutant channels recover two times faster than WT channels. All gating parameters are listed in Table 2.
As a result of the depolarizing shift of steady-state inactivation, the window currents of mutant L1624P channels were increased by 27.73% (Figure 2(c) inset). Therefore, we expected an increase of persistent sodium currents. To test this hypothesis, we examined currents that were elicited by 100 ms depolarizing pulses between −40 and 10 mV (in 10 mV increments) and calculated the ratio of the non-inactivating current, which could be blocked by tetrodotoxin (TTX) (data not shown), and the peak transient sodium current at various potentials (Figure 3 inset). We found a significant increase of this ratio for test potentials near threshold of an action potential in cells expressing mutant L1624P channels (0.17 ± 0.02) compared with WT-expressing cells (0.08 ± 0.03) (Figure 3). Altogether, these results of an increased window and persistent current as well as an accelerated recovery from fast inactivation indicate a gain of function and predict an increase in neuronal excitability via depolarization of the membrane and a shortening of the refractory period after an action potential.
Persistent sodium currents of WT and mutant L1624P channels.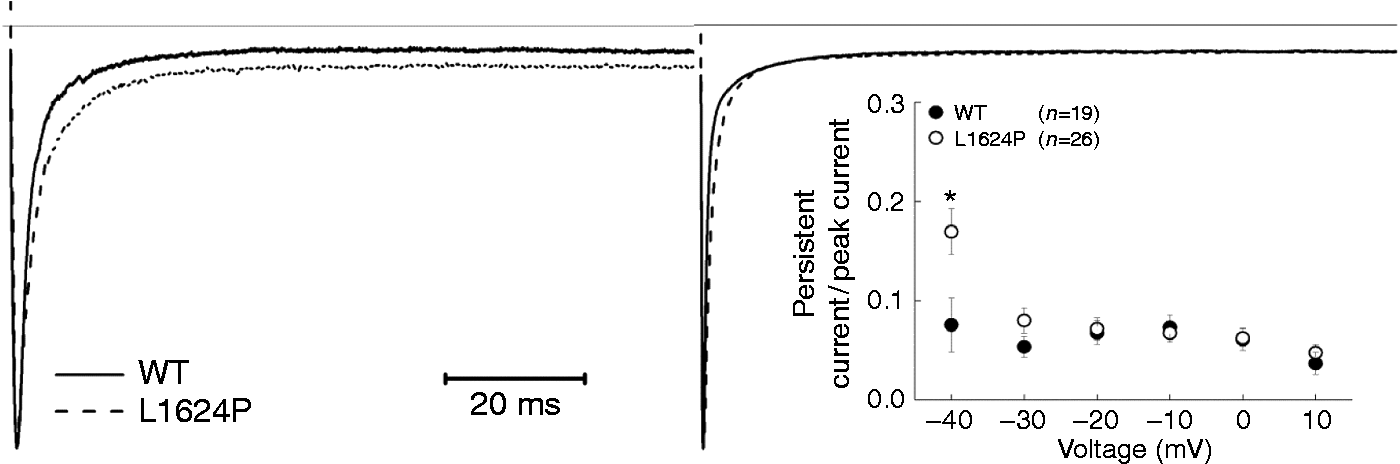
We also tested the slow inactivation properties both for WT and mutant channels (Figure 4). Entry into slow inactivation was monoexponential, with a time constant around 3000 ms for both WT and L1624P channels without significant difference (Table 2). There was no voltage shift of steady-state slow inactivation for L1624P compared to WT, while the slow inactivation voltage sensitivity was significantly different (Table 2). Both WT and L1624P recover in a biexponential way from slow inactivation without significant differences (Table 2). Compared to WT, the slow inactivation properties of L1624P almost remained unchanged.
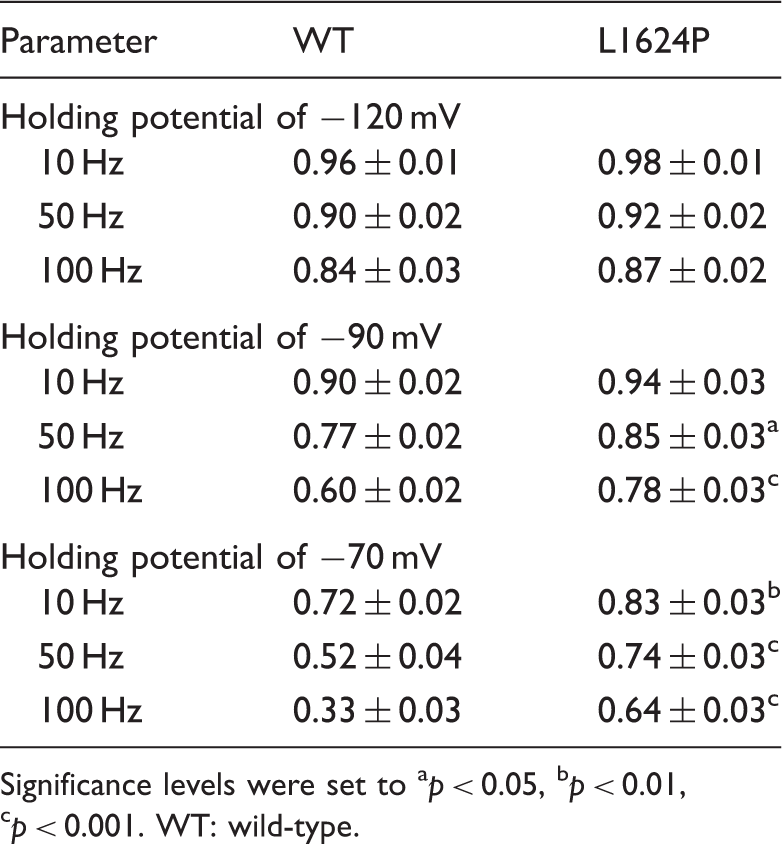
To simulate the channel availability during neuronal firing, we recorded the so-called use-dependence, i.e. whole-cell peak currents during 300 repetitive depolarizing voltage steps to −10 mV at different frequencies and from different holding potentials (Figure 5). At the holding potential of −120 mV, there was no significant difference between the availability of WT and L1624P channels, with both maintaining high channel availability. However, at holding potentials in the physiological range (i.e. more depolarized), mutant channels exhibited a significantly higher availability compared with WT channels. Especially around the neuronal resting membrane potential of −70 mV and a stimulation frequency of 100 Hz (Figure 5), the residual channel availability of mutant L1624P channels was 0.64 ± 0.03, almost twofold higher than for WT channels (0.33 ± 0.03), indicating a marked gain of function in the physiologically relevant range (Table 3). Altogether, our functional studies show a clear gain-of-function effect for mutant NaV1.1 in comparison to WT channels.
Slow inactivation properties of WT (filled circles) and mutant L1624P (open circles). Use dependence of WT and mutant L1624P channels. Parameters of use dependence. Significance levels were set to ap < 0.05, bp < 0.01, cp < 0.001. WT: wild-type.

Discussion
Here we report a German family with pure FHM. In detail, there is a high intra-individual clinical variability with respect to attack duration, severity and frequency. Maybe the most notable clinical feature is that all patients were found to have an early onset of the attacks before puberty (mean age 7 (range 5 to 8) compared to a mean age of 13 in other reported FHM3 families (range 6 to 24) (24)). Permanent neurological disturbances that have been previously described for FHM1 and 2 families such as cognitive deficits (FHM1 and 2) or cerebellar dysfunction (FHM1) (25,26) were not observed. In addition, we did not observe elicited repetitive daily blindness, for which a partial cosegregation with FHM had been described in two French FHM3 pedigrees (19). The identified novel mutation, expanding the spectrum of FHM3 mutations by increasing their number to nine, is located in the S3 segment of domain IV, in the same domain as two of four known mutations causing pure FHM (F1661L and L1649Q). Until now, mutations causing pure FHM3 have been reported to affect this domain as well as the DIII/IV linker, whereas the others are located in different regions of the channel (see also Figure 1), therefore the novel L1624P mutation expands the spectrum of FHM3 mutations by increasing their number to nine. Other missense mutations in IV/S3 have been described causing different clinical phenotypes such as severe myoclonic epilepsy of infancy (SMEI), borderline SMEI (SMEB) or intractable childhood epilepsies with frequent generalized tonic-clonic seizures (27–30).
Functional studies of L1624P revealed a severe disturbance of fast inactivation with a slower inactivation time course, an 11-mV depolarizing shift of the steady-state inactivation curve, and a two times accelerated recovery from fast inactivation. These effects suggest a destabilization of the inactivated state. L1624P substitutes a proline residue with a high turn propensity (31) for the hydrophobic, relatively long side-chain of leucine; L1624P therefore is likely to introduce a hinge in the S3 segment of domain IV distorting the regular structure of the segment (32). Segment IV/S3 has been shown to be involved in fast inactivation before (as, for example, suggested for the muscle sodium channel NaV1.4; (33)). It is therefore conceivable that L1642P affects the channel’s fast inactivation.
Also mutant L1624P channels displayed an abnormal threshold-near persistent current that might be due to the increased window current. This is different from the persistent current detected in other FHM3 mutations, which appear even at more depolarized potential ranges (11,21,22). This unusual persistent current near the threshold might also contribute to more sodium influx during the repolarizing phase of action potentials and facilitate the longer duration of action potentials leading to increased neuron excitability (21). Investigation of slow inactivation properties showed L1624P mostly unchanged, which indicates slow inactivation might not be involved in the pathogenesis of FHM.
All these results indicate a clear gain of function of mutant NaV1.1 channels predicting increased availability and enhanced sodium influx. Furthermore, the higher availability maintained by mutant L1624P in use-dependence experiments suggests that neurons expressing L1624P mutant channels can sustain high-frequency firing better than cells expressing WT channels, or that cells with mutant channels can generate higher firing frequencies than neurons with only WT channels. This is in agreement with most previous results of functional studies of FHM3 mutations (5,11,17,22) and strengthens the main hypothesis, that gain-of-function mutations in the SCN1A gene cause FHM3. As NaV1.1 is the predominant channel in GABAergic interneurons (13,14), our results are compatible with increased firing of inhibitory neurons. And increased firing of inhibitory GABAergic neurons may lead to higher extracellular potassium concentrations, which could trigger CSD as the neurophysiological correlate of the migraine aura (11).
In summary, we add the ninth mutation L1624P to the spectrum of FHM3 mutations. The unusual earlier onset might at least partly be related to this novel mutation. The gain-of-function defects, including the unique persistent current appearing near the threshold, are consistent with the pathomechanisms underlying FHM3. Here, we provide a detailed description of the novel L1624P mutation from the clinical, genetic and molecular points of view that will contribute to the understanding of the pathogenesis of the underlying migraine.
Article highlights
Our studies identify a novel L1624P mutation in NaV1.1 linked to pure familial hemiplegic migraine 3 (FHM3) with a remarkably early age of onset. Our gain-of-function results, including the unique threshold-near persistent current, are consistent with hyperexcitability of inhibitory interneurons, which, by means of increasing extracellular potassium, may facilitate cortical spreading depression (CSD).
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Frank Lehmann-Horn and Karin Jurkat-Rott are fellows of the non-profit Hertie Foundation.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the German Federal Ministry of Research, BMBF and the IonNeurONet (grant number 01GM1105B).