
Abstract
Background
Migraine is a common disabling condition that affects approximately 15% of the population. Several genome-wide association studies have attempted to identify susceptibility variants involved in migraine, reporting several candidate loci for the disorder.
Methods
In order to replicate findings from previous genome-wide association studies, a case–control association study was performed. Twelve single nucleotide polymorphisms were genotyped in a Spanish sample of 512 migraine with aura patients and 535 migraine-free controls.
Results
Nominal associations were found for single nucleotide polymorphisms rs2651899 (within the PRDM16 gene), rs10166942 (near TRPM8), rs12134493 (close to TSPAN2) and rs10504861 (near MMP16) in our migraine with aura sample.
Conclusions
Our study provides suggestive replication, in a Spanish migraine with aura sample, of four genome-wide association study findings previously reported in common migraine. However, larger sample sets should be explored to confirm our results.
Introduction
Migraine is a disabling complex neurological disorder that manifests with episodic and recurrent attacks, affecting around 15% of the population. It is more prevalent in women than men (three to one female:male ratio) (1). Genetic factors are thought to play an important role in common migraine, although environmental influence is also relevant (2).
Two clinical forms of the disorder are distinguished (International Classification of Headache Disorders, ICHD): migraine with aura (MA) and migraine without aura (MO) (3). Both are characterized by the presence of recurrent headaches associated with photophobia, phonophobia, nausea and aggravation by physical activity and are differentiated by the presence of aura. Aura symptoms, which often precede and accompany the headache episode, develop gradually and are most commonly visual but can be sensory, motor, or any combination of these, and can last from a few minutes to several days, although they typically abate in less than 60 minutes (4). MO is the most common migraine presentation, whereas MA is found in about one-third of migraine patients and is thought to have a stronger genetic load (4).
Association studies have been carried out to identify susceptibility variants underlying common migraine at two levels: analysis of candidate genes and genome-wide association studies (GWAS). Several GWAS have been performed so far, four of which reported significant results, disclosing a total of 20 associated variants (1,4–7).
The first risk variant identified, rs1835740, was revealed by a two-stage clinic-based study performed by the International Headache Genetics Consortium (IHGC) in individuals from Europe (5), the first stage consisting of a clinic-based MA sample. Although replication attempts have failed to find association in other populations (1,6,8), this variant is likely to be functional and is located close to the MTDH and PGCP genes, both related to glutamate homeostasis. Additionally, three new genome-wide significant loci were reported in a second GWAS, carried out by Chasman et al. (1) in a population-based study from the Women’s Genome Health Study: PRDM16, LRP1 and a variant located close to TRPM8, all of them identified when the discovery and the replication cohorts (the Dutch Genetic Epidemiology of Migraine study, the German Study of Health in Pomerania and the IHGC discovery stage cohort used in the first migraine GWAS) were combined.
In contrast, no genome-wide significant associations were found in a meta-analysis of population-based migraine GWAS performed by the Dutch-Icelandic Migraine Genetics Consortium and including six European cohorts (6).
More recently, the IHGC carried out a clinic-based GWAS focused on MO patients and reported 12 nominally significant signals in the discovery stage (p-value <10 e-5) (7), two of which, located in the LRP1 gene and close to TRPM8, had already been reported in a previous GWAS (7).
Finally, a GWAS meta-analysis was performed on 29 clinic- and population-based studies including 23,285 migraine cases and 95,425 population-matched controls (4). Association with 12 variants within or close to the following genes were reported: PRDM16; MEF2D; TRPM8; TGFBR2; PHACTR1; ASTN2; LRP1; FHL5; C7orf10; MMP16; TSPAN2; AJAP1. The last five associations were reported in this study for the first time.
Here we performed a clinic-based case–control association study in 512 MA patients and 535 non-migraine individuals from Spain aiming at replicating significant associations identified in those previous GWAS that included MA patients (1,4), although they also included MO individuals. The other three GWAS were not considered here because one of them did not render positive findings (6), another one included only MO patients (7) and the third one (5) was already subjected to replication by us in a previous report (8).
Materials and methods
Patients and controls
Clinical findings in 517 patients with migraine with aura included in this study.

MA: migraine with aura.
The percentage of available answers per item ranged from 88.3 to 99.2%.
In first degree relatives.
Mean ± SD.
DNA isolation, SNP selection and genotyping
DNA was extracted from peripheral blood lymphocytes by a standard salting-out procedure (9).
Selected SNPs for replication of GWAS findings. Comparison of the direction of the effect for the nominally associated SNPs in our study.

MA: migraine with aura; MO: migraine without aura; SNP: single nucleotide polymorphism.
Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly.
Genotyping failed in our study.
Excluded from our study (controls not in Hardy–Weinberg equilibrium).
Additional association with the ‘MO only' group.
Additional association with the ‘clinics only (MA and MO)' group.
Additional association with the ‘All samples' group.
Minor allele frequencies (MAFs) and odds ratios (ORs) are expressed as a range for the study of Chasman (performed in different population groups).
Minor allele in ( + ) strand.
Genotyping of all SNPs was performed using the Sequenom technology at the National Genotyping Center (Cegen, Santiago de Compostela, Spain). Three samples from the Centre d'Étude du Polymorphisme Humain (CEPH) were included in every plate and a 100% concordance with HapMap data was observed when available. One SNP, rs2274316 in MEF2D, failed and it was not considered in the association study. Five patients displayed less than 85% genotyping rate and were removed from the study, so the analysis was finally carried out on 512 patients.
Statistical analysis
The analysis of minimal statistical power for the x2 test, performed under a dominant model using the Genetic Power Calculator software (pngu.mgh.harvard.edu/∼purcell/gpc) with α = 0.05, an odds ratio (OR) of 1.25, a prevalence of 0.15 for the disorder and the highest minor allele frequency (MAF) found in controls (0.455), rendered a value of 47% in the overall sample.
Statistical analysis for the association study was carried out using the SNPassoc R library (10). Hardy–Weinberg equilibrium (HWE) was tested in our control sample, and P-values were all over 0.05, except for rs10915437 (P = 0.0470), which was not further considered in our analysis. We compared allele and genotype frequencies using x2 tests and considering co-dominant, dominant, recessive, overdominant and log-additive models. For the log-additive model the Mantel–Haenszel x2 test was implemented. All the results were adjusted by sex using a likelihood ratio test. Corrections for multiple testing were applied using 10,000 permutations with PLINK (11) and Bonferroni’s correction. For the latter, the threshold for significance was established as 0.0003 = 0.05/(11 × 3 × 5), with 11 SNP tests, three clinical groups and five genetic models.
Association between case–control status and the different genetic variants was also estimated using logistic regression and including sex as a predictive variable (logit = βo + β1*SNP + β2*sex). Analyses assuming additive, dominant, recessive and co-dominant models for each SNP were performed separately and then compared using Akaike Information Criterion (AIC). All the analyses were performed with the R software (http://CRAN.R-project.org).
Results
Case–controls association study performed in our sample of 512 MA patients and 535 control individuals.
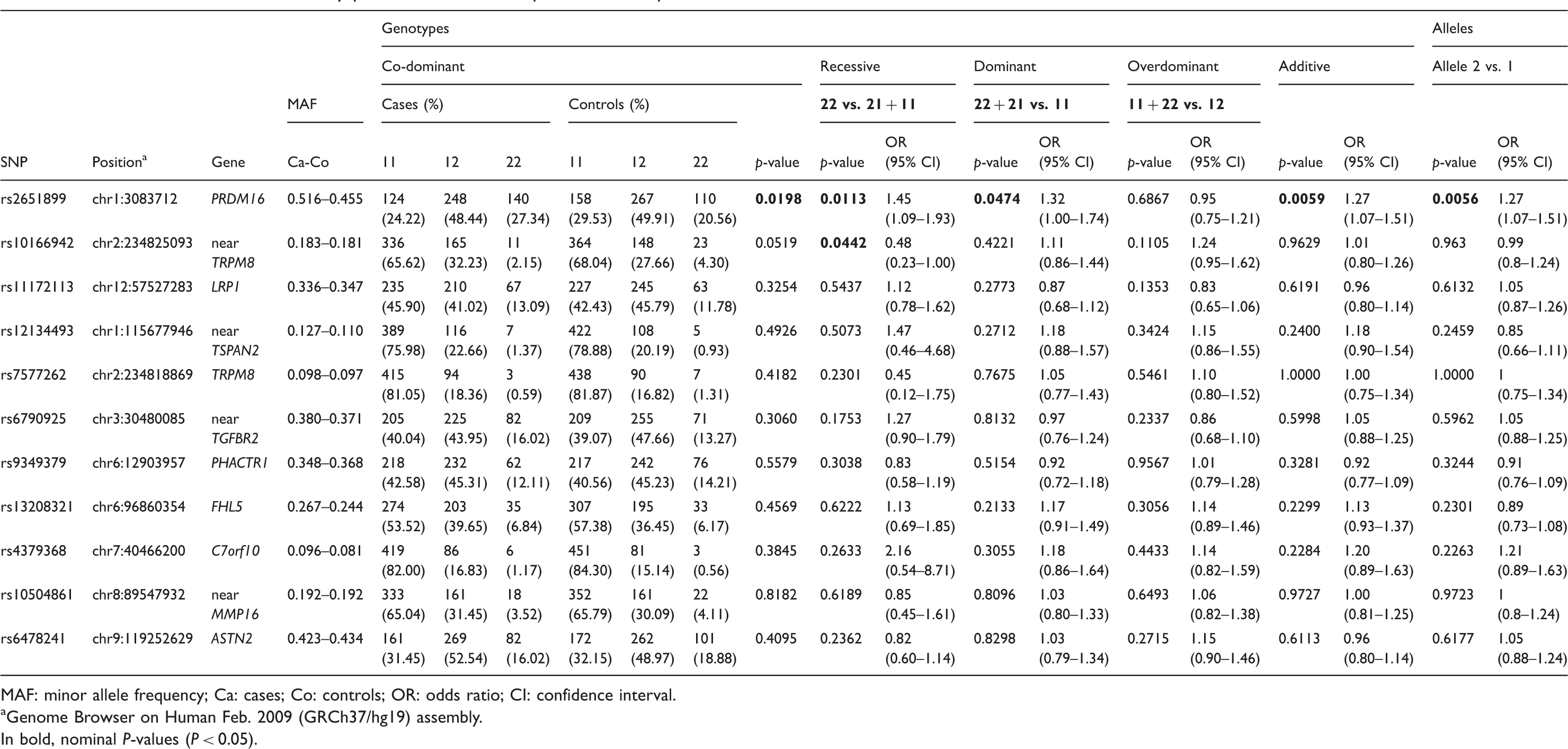
MAF: minor allele frequency; Ca: cases; Co: controls; OR: odds ratio; CI: confidence interval.
Genome Browser on Human Feb. 2009 (GRCh37/hg19) assembly. In bold, nominal P-values (P < 0.05).
We achieved nominal associations for two out of three genome-wide significant findings from the study by Chasman et al. (1): rs2651899 in the PRDM16 gene (allelic model, P = 0.0056) and rs10166942, located near TRPM8 (recessive, P = 0.0442) (Table 3). However, none of the SNPs survived Bonferroni’s correction for multiple testing (all P-values > 0.0003) or permutation corrections (all permuted P-values > 0.05, data not shown). The evaluation of the remaining nine SNPs, selected from the meta-analysis study performed by Anttila et al. (4), failed to replicate any of the associations in our MA sample.
We subsequently split the sample into the subclinical groups ‘MA only’ and ‘Both MA and MO’ to gain clinical homogeneity at the expense of reducing the size of the sample, following the subdivision reported in Anttila et al. (4). Only one of the previous association signals remained significant (rs2651899, ‘MA only’, allelic model, P = 0.0034) but novel nominal associations were obtained with ‘Both MA and MO’ for SNPs rs12134493, located near TSPAN2 (dominant model, P = 0.0362) and rs10504861, near MMP16 (recessive model, P = 0.0288) (see Table 1 in online Supplementary Material).
We also tested association between the SNPs and the different phenotypes using logistic regression modelling, that rendered significant results for SNPs rs2651899 (‘All MA', additive, P = 0.0061; ‘MA only', additive, P = 0.0038) and rs12134493 (‘Both MA and MO', additive, P = 0.0417), in all cases with the additive model as the most suitable one for predicting the disease status (AIC values of 1446.7, 1123.8 and 804.14, respectively). However, non-significant results were obtained for rs10166942 and rs10504861 (see Table 2 in online Supplementary Material).
In all cases the direction of the effect was consistent with the previous reports (Table 2). Specifically, for rs2651899 and rs12134493 the risk allele is the minor one both in the original report and in our replication study, C and A in the (+) DNA strand, respectively. In contrast, for rs10166942 and rs10504861, the associated risk alleles, T and C, are the most frequent ones in all studies.
Discussion
In summary, four out of 12 tested SNPs revealed a nominal association with MA in our sample, tentatively replicating previous GWAS findings: rs2651899 (PRDM16), rs12134493 (near TSPAN2), rs10166942 (near TRPM8) and rs10504861 (near MMP16). The first two associations are the most robust replication signals in our study, as they emerge in the x2 testing under different genetic models and in the logistic regression analysis. In contrast, rs10166942 and rs10504861 show association only under the recessive model in the x2 comparisons and not in the logistic regressions.
SNP rs2651899 was found to be associated with migraine in the two previous studies that we aimed to replicate (1,4) and more recently in a replication study carried out in a Chinese sample (12) and thus emerges as the most consistent hit across this set of GWAS studies. rs2651899 is located in the first intron of the PRDM16 gene and its role in the pathophysiology of migraine remains still unclear. However, it has recently been associated with cardiac disease (non-syndromic left ventricular non-compaction cardiomyopathy and dilated cardiomyopathy) (13). Given the known co-morbidity between cardiovascular disease and migraine, PRDM16 may be considered as a candidate gene in future studies. The association with SNP rs10166942 has previously been replicated in a Danish population (2). This variant is located close to TRPM8, a gene that has been consistently associated with migraine in several studies, supporting a connection between migraine and neuropathic pain (1). Our study is the first to replicate the association of rs12134493 and rs10504861 with migraine, with nominal associations found in our ‘Both MA and MO’ subgroup. rs12134493 lies next to TSPAN2, a gene encoding a cell surface protein, which mediates signal transduction. rs10504861 is located near MMP16, encoding a matrix metalloproteinase (MT-MMP2/MMP16) responsible for the cleavage of LRP1, a protein known to increase axonal and synaptic plasticity in cortical neurons. A SNP in the LRP1 gene, rs11172113, also showed association with migraine in a GWAS (1,4) and in a subsequent replication study (2), but displayed negative results in our study.
There are coincidences and discrepancies between our results and the associations reported previously, which may be explained by several methodological issues: (i) the sample size of our cohort (about 500 patients and 500 controls) is smaller than those of the previous GWAS. Thus, the study is underpowered and the likelihood of false negative findings is high. Although we found nominal findings, none of them survived multiple testing corrections; (ii) our patients' group consists of subjects clinically diagnosed with MA, while the previous studies that we are replicating include MA and also MO patients. Although MA and MO share clinical features, their genetic background may differ, at least in part, as shown by previous studies (4). To shed light on the shared and specific genetic risk factors for MA and MO, it would be interesting to systematically cross-test any migraine-associated SNP in the two clinical entities. Moreover, the fact that we are comparing clinic- and population-based studies may be another reason for the discordant results, as diagnosis of aura is usually more reliable in a clinical setting; (iii) in our sample, the proportion of pure MA differs from that reported in previous studies. Likewise to the MO vs. MA paradigm, it is conceivable that sub-phenotypes of MA (i.e. ‘MA-only’ vs. ‘Both MA and MO') may conceal specific genetic backgrounds; (iv) the small number of positive replications in our study may also be related to the underlying architecture of MA vs. MO. It is conceivable that rare variation plays a more important role than common variation in MA, whereas frequent variants would be more relevant in MO. In favour of this hypothesis is that most association findings from GWA studies have been identified in MO or in ‘all migraine' samples.
In conclusion, although our results do not attain statistical significance, the nominal findings obtained here suggest that previously reported loci may be involved in the genetic architecture of MA. However, additional replication studies in larger MO and MA sets are warranted to weigh up the relevance of these susceptibility loci to both common migraine phenotypes.
Article highlights
Case–control association study of 512 migraine with aura patients and 535 migraine-free controls aimed at replicating previously reported migraine GWAS findings. Suggestive association with four out of 12 loci studied: one SNP in the PRDM16 gene and three SNPs located close to TRPM8, TSPAN2 and MMP16. These genes have been related to cardiac disease, neuropathic pain, signal transduction or axonal/synaptic plasticity.
Footnotes
Funding
Funding for this study was provided by the Spanish Ministry of Economy and Competitiveness (SAF2009–13182-C01, SAF2009–13182-C03), AGAUR (2014SGR-0932, 2009SGR-0078), Fondo de Investigaciones Sanitarias (FIS: PI10/00876) and Fundació La Marató de TV3 (072310). These institutions had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Conflict of interest
P.P.R. reports support for conference visits from MSD and Almirall, and consultancy for Allergan. A.M. received honorary fees for lectures and advisory boards from Novartis Europe.
Acknowledgments
We thank all the patients who participated in this study.