
Abstract
Background
Recent studies evaluated short-term efficacy and safety of peripheral nerve stimulation (PNS) of the occipital nerves for managing chronic migraine. We present 52-week safety and efficacy results from an open-label extension of a randomized, sham-controlled trial.
Methods
In this institutional review board-approved, randomized, multicenter, double-blinded study, patients were implanted with a neurostimulation system, randomized to an active or control group for 12 weeks, and received open-label treatment for an additional 40 weeks. Outcomes collected included number of headache days, pain intensity, migraine disability assessment (MIDAS), Zung Pain and Distress (PAD), direct patient reports of headache pain relief, quality of life, satisfaction and adverse events. Statistical tests assessed change from baseline to 52 weeks using paired t-tests. Intent-to-treat (ITT) analyses of all patients (N = 157) and analyses of only patients who met criteria for intractable chronic migraine (ICM; N = 125) were performed.
Results
Headache days were significantly reduced by 6.7 (±8.4) days in the ITT population (p < 0.001) and by 7.7 (±8.7) days in the ICM population (p < 0.001). The percentages of patients who achieved a 30% and 50% reduction in headache days and/or pain intensity were 59.5% and 47.8%, respectively. MIDAS and Zung PAD scores were significantly reduced for both populations. Excellent or good headache relief was reported by 65.4% of the ITT population and 67.9% of the ICM population. More than half the patients in both cohorts were satisfied with the headache relief provided by the device. A total of 183 device/procedure-related adverse events occurred during the study, of which 18 (8.6%) required hospitalization and 85 (40.7%) required surgical intervention; 70% of patients experienced an adverse event.
Conclusion
Our results support the 12-month efficacy of PNS of the occipital nerves for headache pain and disability associated with chronic migraine. More emphasis on adverse event mitigation is needed in future research. Trial registration: Clinical trials.gov (NCT00615342).
Introduction
Chronic migraine (CM), a neurological disorder that impacts approximately 2.0% of the general population, imposes a substantial economic burden on society and is associated with significant disability and impairment in quality of life (1–6). CM has only recently been defined, and the definition and endpoints to be evaluated in clinical trials continue to evolve (7–11). There have been relatively few controlled clinical trials evaluating the safety and efficacy of treatments for CM. A large unmet medical need exists for this prevalent and disabling condition.
Open-label, controlled clinical trials evaluated the safety and efficacy of peripheral nerve stimulation (PNS) in the occipital region for the treatment of CM (12–17). Although the results from open-label studies reported promising results, the results from controlled trials have been mixed. In the small feasibility ONSTIM (Occipital Nerve Stimulation for the Treatment of Chronic Migraine Headache) study, 39% of patients in the adjustable stimulation group experienced at least a 50% reduction in headache frequency or a fall of at least 3 points on the intensity scale, compared with 6% in the sham-stimulation and 0% in medically managed groups (16). In the Precision Implantable Stimulator for Migraine (PRISM) study, 139 patients with episodic and chronic migraine were randomized to receive either active bilateral stimulation or sham stimulation only if a preceding 5–10 day stimulation trial with external leads was successful (17). Occipital nerve stimulation did not produce significant benefits in relation to sham stimulation for the change from baseline in migraine days per month evaluated 12 weeks after implantation. Subgroup analysis identified several candidate predictors of a favorable response, including the absence of medication overuse, no use of opioids and a positive response to a trial of percutaneous stimulation (17). In those randomized to the active group who derived a positive response to trial stimulation compared with those who had no response during the trial stimulation period, the reduction in migraine days was 8.8 compared with 0.7 (p < 0.001).
In a larger prospective, randomized, controlled, multicenter study, 157 patients implanted with a neurostimulation system were evaluated at 12 weeks (18). There was no significant difference between the active and control groups in the percentage of responders reaching a 50% reduction in mean daily visual analogue scores (VAS) at 12 weeks. However, there were significant reductions in pain intensity, headache days and migraine-related disability. Although these results provide a level of optimism for the potential of this treatment modality, the invasive nature and cost associated with the procedure demand that long-term outcomes be accurately defined for patients, as well as regulatory and reimbursement authorities. We report the 52-week safety and efficacy outcomes of patients enrolled into this study.
Materials and methods
Sample size
Methods for determining sample size were described previously and based on the controlled phase of the study (18). Briefly, a low response rate was expected in the control group as a result of the primary endpoint requirement of 50% reduction in mean daily VAS scores for average pain. The response rate was assumed to be 15% and the population response rate among patients in the active group was assumed to be 45%. Based on these estimates, 150 patients randomized 2:1 (100 in the active group and 50 in the control group) provided 80% power to reject the null hypothesis at a significance level of α = 0.05.
Participants and study design
Key inclusion/exclusion criteria.

The United States Food and Drug Administration approved the study protocol, the study was reported on clinicaltrials.gov (NCT00615342), and all sites received institutional review board approval prior to study initiation.
Surgical procedure
Implantation of the permanent PNS system occurred only in patients who underwent a successful trial (defined as at least 50% reduction in pain or adequate paresthesia coverage in the painful areas) to determine proper lead placement. Patients who did not have a successful trial (n = 20) were classified as screen failures and were exited from the study. A total of 157 patients were implanted from November, 2005 to June, 2009. The implant procedure has been described previously (18). Briefly, patients had percutaneous quadripolar leads (Quattrode, St. Jude Medical, Plano, TX, USA) placed on either side of the midline perpendicular to the course of the occipital nerves (n = 126) at the level of the craniocervical junction, or less commonly craniocaudally parallel to the course of the nerve (n = 24), oblique (n = 7). Depending on the pain distribution, leads were placed either unilaterally (n = 7) or bilaterally (n = 150). The Genesis (St. Jude Medical, Plano, TX, USA) non-rechargeable implantable pulse generator (IPG) was implanted in a subcutaneous pocket created so the IPG was parallel to and not more than 4 cm (1.5 in) below the skin surface. The lead or extension was tunneled subcutaneously to the pocket, connected to the IPG and the incisions were closed.
Randomization and masking
Randomization and masking during the controlled phase of the study were described previously (18). After permanent implant, patients were randomized into either an active (n = 102) or control group (n = 52) in a 2:1 ratio using a block size of three (SAS version 9.2). The 2:1 randomization was chosen to minimize the number of patients who received no stimulation, and to maximize the number of patients who received active stimulation for determination of device/procedure-related adverse events (AEs). Two sets of sealed envelopes were provided to each investigator and the appropriate set was opened by a sponsor representative. Both investigators and patients were blinded to treatment.
Programming
Patients in the active group were programmed for appropriate stimulation, whereas patients in the control group were given a sham programmer that did not communicate with the IPG. At the conclusion of the 12-week, controlled phase of the study, all patients received active stimulation for the remainder of the study. Given the delayed device activation in the control group, patients in that group received stimulation for 40 weeks at the conclusion of the 52 week study, whereas patients in the active group received stimulation for 52 weeks. Patients were permitted to use established pain medications and other treatment modalities already in use 8 weeks prior to baseline at the same levels, but new methods of pain control were prohibited.
Data collection
The primary outcome for the controlled phase of the study was mean daily VAS measurements recorded in a patient diary, and the primary endpoint was a comparison of the proportion of responders in the active group with those in the control group at 12 weeks. Results for the primary outcome and endpoint were reported elsewhere (18); here we report secondary outcomes of the study for the open-label extension. The primary aims for this analysis included the reduction in number of headache days (duration > 4 hours with peak intensity reported as moderate or severe), percentage of patients who achieved a 30% or greater or a 50% or greater reduction in headache days and/or pain intensity as measured by a VAS. Secondary aims for this study included migraine-related disability and distress as assessed by the migraine disability assessment (MIDAS) questionnaire and the Zung Pain and Distress (PAD) scale, direct patient reports of headache pain relief (categorical and percentage), quality of life (3 point categorical rating of improved, stayed the same or deteriorated), satisfaction (dichotomous rating of yes or no) and AEs.
Questionnaires were administered during scheduled follow-up visits at 24 and 52 weeks after permanent implant. Electronic patient diaries were used to collect headache information including presence, average and peak intensity, duration, symptoms and medication use. Diary data were collected during the 4 weeks preceding the baseline assessment, and again during the 4 weeks preceding the 52-week visit (between weeks 48 and 52). If a patient completed less than 14 diary days during the assessment period, the patient's value was missing for that period.
An intent-to-treat (ITT) patient group included all patients (N = 157), and a patient group of those who met criteria for intractable chronic migraine (ICM; N = 125) were analyzed for all variables during the open-label phase of the study. The ICM population included patients that met the criteria for chronic migraine, had failed three or more preventive drugs and were at least moderately disabled (MIDAS score of ≥ 11) at baseline.
Statistical methods
All statistical analyses were performed using SAS version 9.2 (Cary, NC,USA). All statistical tests were two-sided with a significance level of 5%, unless otherwise specified. Missing data varied as some patients were missing certain measures and scores, thus only patients with available data were included in each analysis. The number of patients with available data is reported for each measure. Initially, data were analyzed to assess change from baseline after 24 or 52 weeks of stimulation in the active group, or after 12 and 40 weeks of stimulation in the control group. Data from both treatment groups were combined at all time-points after the 12-week controlled phase of the study, as analysis showed that data for patients in the control group did not differ from data for patients in the active group despite patients in the control group receiving stimulation for a shorter duration of time.
Reduction in the number of headache days (duration > 4 hours with peak intensity reported as moderate or severe) from the baseline period to 52 weeks (normalized to 28 days), change in MIDAS and Zung PAD scores at 24 (where applicable) and 52 weeks were analyzed using paired t-tests. We hypothesized that headache days, MIDAS and Zung PAD scores would be significantly decreased from baseline by PNS of the occipital nerves at all time-points analyzed. Direct patient report of pain relief (categorical classification and percentage), quality of life and satisfaction were summarized and are presented. For AEs, data are presented as the number and percentage of all AEs.
Results
A total of 268 patients were enrolled from 15 investigational sites between 30 June 2005 and 20 August 2010, and 157 were implanted with a permanent system and randomized (Figure 1). Patient demographics and baseline characteristics are presented in Tables 2 and 3.
Patient disposition. Patient demographics. Patient baseline characteristics.

Reduction in headache days
Reduction in headache days at 52 weeks is displayed in Figure 2. Headache days data at baseline and 52-week were available for 111 patients in the ITT population and for 89 patients in the ICM population. Mean (±SD) headache days at baseline were 21.6 (±7.1) for the ITT population and 24.2 (±4.5) for the ICM population. At 52 weeks post-implant, mean headache days were significantly reduced from baseline by 6.7 (±8.4) in the ITT population and by 7.7 (±8.7) in the ICM population. Reduction in headache days was significant for both the ITT (p < 0.001) and ICM populations (p < 0.001). The percentage of patients who achieved a 30% reduction in headache days and/or pain intensity as measured by a VAS was 59.5%, and the percentage who achieved a 50% reduction was 47.8%.
Headache days were significantly reduced by 6.7 (±8.4) days (p < 0.001) in the ITT population and by 7.7 (±8.7) days (p < 0.001) in the ICM population. After week 12, all patients (active and control) were collapsed into one group. Error bars represent standard deviations.
MIDAS questionnaire
Initially, the differences in MIDAS scores were analyzed for patients who had only experienced 12 weeks of stimulation (those initially assigned to the control group) compared with patients who had experienced 24 weeks of stimulation (those assigned to the active group at study start) in the ITT cohort at the 24 week time-point. This analysis showed no significant differences in MIDAS scores between these two groups. The patients who received only 12 weeks of stimulation achieved MIDAS scores comparable with patients who received 24 weeks of stimulation, indicating no differences between active and control groups by the 24-week study visit. Thus, for the focus of this study, these two groups were combined into one group to determine any change in MIDAS scores from baseline scores.
The mean baseline MIDAS score for the ITT population (n = 142) was 156.6 (±75.3) points, which was significantly reduced by 50.9 (±62.8) points (p < 0.001; n = 140) at 24 weeks and by 50.9 (±71.9) points (p < 0.001; n = 133) at 52 weeks. For the ICM population (n = 114), MIDAS scores were significantly reduced by 56.9 (±63.4) points (p < 0.001; n = 112) at 24 weeks and by 57.9 (±71.8) points (p < 0.001; n = 106) at 52 weeks from a baseline score of 169.7 (±70.6). Results are depicted in Figure 3.
In the ITT population, MIDAS scores were significantly reduced by 50.9 (±71.9) points to a mean score of 106.7 (±85.4) points (p < 0.001), and in the ICM population scores were significantly reduced by 57.9 (±71.8) points from a baseline score of 169.7 (±70.6) points (p < 0.001). After week 12, all patients (active and control) were collapsed into one group. Error bars represent standard deviations.
Zung PAD scale
Total scores, as well as pain, mood and behavior subcomponent scores for the PAD scale are shown in Figure 4. For the ITT population (n = 132), total PAD scores were significantly reduced by 10.3 (±14.8) points (p < 0.001) at 52 weeks from a mean baseline score of 66.8 (±13.6) points. Mean baseline total score for the ICM population (n = 105) was 68.6 (±13.3) points, which was significantly reduced by 11.2 (±15.2) points to a score of 57.4 (±16.2) points at 52 weeks post-implant. The pain subcomponent score was significantly reduced by 9.0 (±27.0) points (p < 0.001) from a baseline score of 78.8 (±25.7) in the ITT population (n = 131) and by 10.8 (±28.0) points (p < 0.001) from a baseline score of 80.5 (±25.4) in the ICM population (n = 104) at 52 weeks. Mood subcomponent scores were significantly reduced in both the ITT population (−12.9 points; p < 0.001; n = 132) and the ICM population (−13.2 points; p < 0.001; n = 105) as were behavior subcomponent scores (−9.2 points in the ITT cohort; p < 0.001; n = 132 and −10.3 points in the ICM cohort; p < 0.001; n = 105).
Total PAD scores were significantly reduced by 10.3 (±14.8) points from a mean baseline score of 66.8 (±13.6) points (p < 0.001) in the ITT population (a) and by 11.2 (±15.2) points to a score of 57.4 (±16.2) points in the ICM population (b; p < 0.001) at 52 weeks. After week 12, all patients (active and control) were collapsed into one group. Error bars represent standard deviations.
Direct patient report of headache relief and quality of life
Again, an analysis was initially performed to rule out active versus control group differences at 24 weeks in the percentage of patients reporting excellent or good headache relief and the percentage of patients reporting improved or greatly improved quality of life in the ITT group. No significant differences existed. Based on the null results, 52-week change from baseline analyses were conducted on combined data.
Patients were asked to categorize their overall headache relief as excellent, good, fair, poor or unable to decide, and to provide the percentage of pain relief. In the ITT cohort, 55.7% (78 out of 140) of patients reported excellent or good headache relief at the 24-week visit and 65.4% (87 out of 133) of the patients reported the same at the 52-week visit. In the ICM population (n = 106), 67.9% of patients reported excellent or good headache relief at the 52-week visit. Less than 20% of patients reported pain relief as poor in either the ITT or the ICM population. Patients in the ITT population reported a mean percentage of pain relief of 43.3% (±29.5) at the 24-week follow-up. At 52 weeks, mean percentage of pain relief was 49.5% (±30.7) in the ITT population and 50.4 (±30.5)% in the ICM population. These results are shown in Figure 5.
Patient categorical rating of headache relief. The majority of patients in both the ITT (65.4%; a) and the ICM (67.9%; b) population reported excellent or good headache relief at 52 weeks.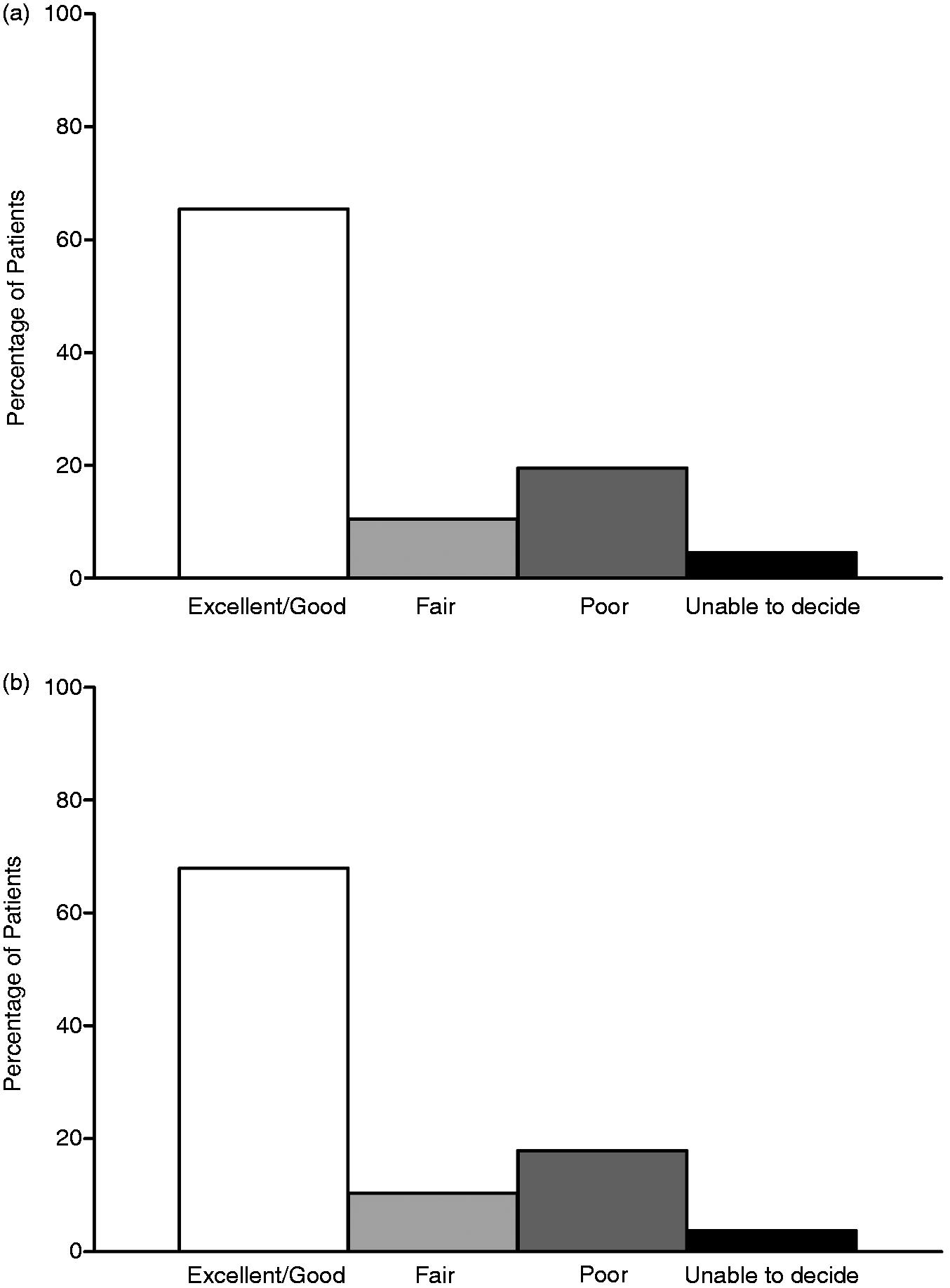
Patients were rated overall improvement in quality of life as improved, stayed the same or deteriorated. At 24 weeks post-implant, 64.3% (90 out of 140) of patients reported improved quality of life in the ITT population. Similar results were noted at the 52-week follow-up with 68.4% (91 out of 133) of patients reporting the same. At 52 weeks post-implant, 69.8% (74 out of 106) of patients in the ICM cohort reported improved quality of life. Less than 10% of patients reported quality of life as deteriorated in both the ITT and ICM population. Results are displayed in Figure 6.
Quality of life. In the ITT population (a), 68.4% of patients reported improved quality of life (n = 133) and 69.8% reported the same in the ICM population (n = 106; b). Less than 10% of patients reported their quality of life as deteriorated in both the ITT and ICM population at 52 weeks.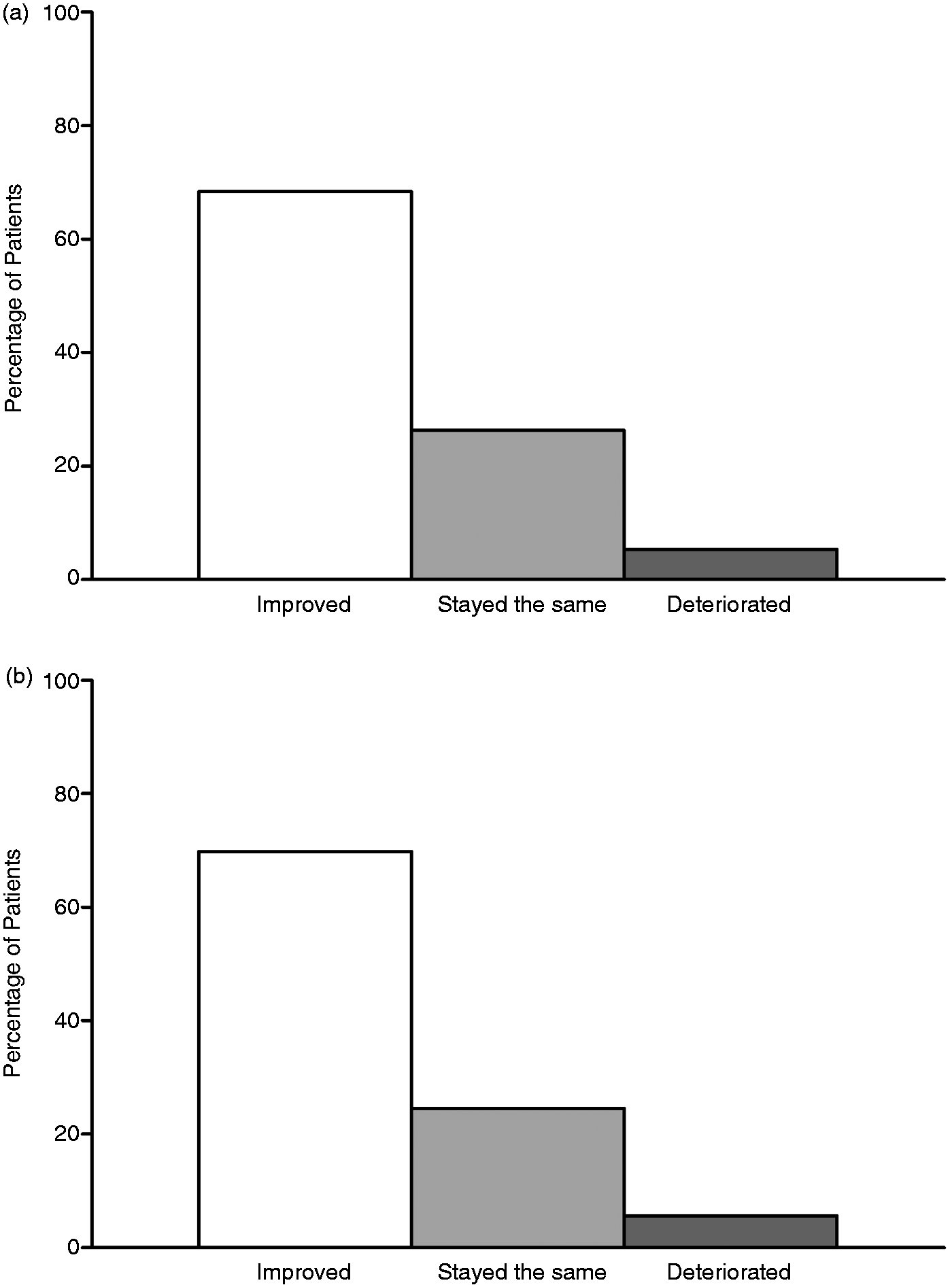
Patient satisfaction
In the ITT group, active and control groups were combined because no significant differences existed at 24 weeks in the percentage of patients reporting satisfaction with the headache relief provided by the device. Patients were asked if they were satisfied with the headache relief offered by the device (yes or no) (Figure 7). Overall, 54.3% (76 out of 140) of patients in the ITT cohort were satisfied with the device at 24 weeks. At 52 weeks, 58.6% (78 out of 133) of patients in the ITT population and 59.4% (63 out of 106) of patients in the ICM population reported the same. Furthermore, 68% of patients in the ITT population (n = 153) and 66.4% of patients in the ICM population (n = 122) reported being very satisfied or satisfied with the results of the procedure. When asked if they would undergo the procedure again, 83.0% of patients in the ITT population (n = 141) indicated yes and 73.0% of patients in the ICM population (n = 122) reported the same. For the ITT population (n = 151), 88.7% indicated that they would recommend the procedure to someone else, and 86.7% of patients in the ICM population (n = 120) indicated the same.
Patient satisfaction. In the ITT population (a), 68% of patients reported being very satisfied or satisfied with the results of the procedure (n = 153) and 66.4% reported the same in the ICM population (n = 122; b) at 52 weeks.
Adverse events
Adverse events.

Data are presented as the number and percentage of all adverse events. All adverse events with an incidence rate of 2% or greater are presented. Overall, the most common hardware-related adverse event was lead migration, which accounted for 13.9% of all events; the most common biological adverse event was persistent pain, and/or numbness at IPG/lead site, which accounted for 18.2% of all events, and the most common stimulation-related adverse event was lack of efficacy or return of symptoms, which accounted for 10.0% of all events. Over 30% (32.5%) of patients did not experience an adverse event.
Other actions taken included hospitalization, injections or nerve blocks, local padding, patient education, programmer replacement, discontinuation of stimulation, removal of sutures and X rays.
There were a total of 209 AEs, and 111/157 (70.7%) of the implanted patients experienced one or more AE. A total of 56 hardware-related events occurred, including eight battery failures, three occurrences of battery passivation, and eight device malfunctions involving lead or extension disconnection (three), extension malfunction (one), programmer malfunction (three) or IPG malfunction (one). There were 29 lead migrations, one IPG migration, and seven lead breakages or fractures. There were 82 biological events, including one subcutaneous hematoma, 11 infections, one seroma, eight skin erosions, five wound site complications, 38 cases of persistent pain and/or numbness at the IPG/lead site, one case of pain or swelling at the IPG site following a car accident, five allergic reactions to surgical materials, 11 cases of expected postoperative pain/numbness at IPG/lead site, and one subcutaneous tissue change at the implant site. Stimulation-related events accounted for 45 cases, including one case of unintended changes in headache severity, type or frequency; 17 cases of undesirable changes in stimulation; 21 cases of lack of efficacy or return of symptoms; one case of unintended stimulation effects-muscle spasms/cramping; four cases of nausea/vomiting; and one case of diminished or loss of motor or musculoskeletal control. The remaining 26 AEs were not considered device- or procedure-related. Of the device/procedure related AEs, 80 (38.3%) were resolved with little to no risk to the patient (i.e. no action taken, reprogramming or medication only), whereas the remaining 18 (8.6%) required hospitalization. Eighty-five (40.7%) AEs, of any type, resulted in an additional surgery. The majority of AEs were classified as mildly or moderately severe, and the rate of serious device- or procedure-related AEs was 10.8% (n = 17). Forty events were classified as serious adverse events, with 23 (58%) of these events considered as non-device or procedure-related.
Discussion
This is the first large-scale, prospective, controlled study evaluating PNS in the occipital region for CM to report 1-year efficacy and safety results. In both the ITT and ICM populations, significant reductions in headache days as well as MIDAS and Zung PAD scores were demonstrated. In addition, almost 60% of patients achieved a 30% or greater reduction in headache days and/or pain intensity as measured by a VAS, and almost half of the patients achieved a 50% reduction. Over two-thirds of patients in each group reported excellent or good headache relief, improved quality of life, and reported being very satisfied or satisfied with the results of the procedure. In the ITT and ICM populations, 83.0% and 73% of patients, respectively, reported that they would undergo the procedure again.
These efficacy results should be interpreted, however, within the context of the safety profile of this therapy. There were 183 device-procedure-related AEs during the course of the study of which 18 (8.6%) required hospitalization, and overall, 85 (40.7%) resulted in an additional surgery The study protocol called for an eight contact system with a large non-rechargeable internal programmable generator. The use of additional contacts may be helpful in reducing the AE of migration, as the addition of more contacts allows for reprogramming to resolve a lead movement issue, rather than surgical revision. The presence of new mechanical anchors and procedures to provide strain relief may also be important in sustaining lead position (19). A recent review of infection reduction by Deer and Provenzano (20) may also lead to changes in clinical practice to reduce infection rates and the need for reoperation. The authors recommend infection prevention measures across all stages of the surgical procedure such as assessment of patient risk prior to the surgical procedure, selection of prophylactic intravenous antibiotics based on known hospital pathogens (preoperative), selection of operating rooms with laminar flow and HEPA filters, limiting surgical time (intraoperative), use of occlusive dressing for a minimum of 24 to 48 hours, and vigilance of tape allergies and skin irritants (postoperative). These changes in methods and protocols should lead to improved safety, while maintaining or improving efficacy.
These results support the previously reported 12-week results (18), and suggest that early benefits are sustained over 12 months. At 12 weeks, there were no significant between-group differences in the number of patients with a 50% reduction on the VAS (primary endpoint). However, there was a significant difference at 30% (p < 0.05), which in this population of patients is considered to be clinically important (10). In addition, significant group differences (p < 0.05) were observed for the reduction in number of headache days, MIDAS, Zung, quality of life and patient satisfaction.
These results support open-label studies that have suggested reasonable long-term outcomes in patients undergoing implantation of PNS in the occipital region for CM. In one retrospective open-label study of 15 patients with chronic headache, of whom eight had CM, headache frequency, disability (MIDAS), depression (Beck II), and headache severity were significantly improved at a mean follow-up period of 19 months (15). The need for surgical revision in this study was high (60%). In other open-label studies, 64% to 100% of patients had improvement rates exceeding 50% after a follow-up period of up to 18 months (12–14).
Despite the preliminary but promising results from this long-term study, some experts remain skeptical about the feasibility, need and future of PNS for patients with CM (21). It is believed that with appropriate multidisciplinary, multimodal and integrated non-surgical care, very few patients with CM have poor outcomes. However, up to 15% of patients with CM remain intractable even after aggressive medical management (22). Concern has been expressed about the cost of care and the high complication rates (23). In a recent study of 27 patients with chronic cluster headache (twenty-four) or CM (three), the per-case cost was 28 186€ (9445€ for hospitalization and 18 741€ for hardware) (23). Although the cost of care is an important consideration, the cost-benefit ratio must be considered in the context of the enormous disability associated with intractable CM, the direct and indirect costs associated with the disorder, and the costs associated with treating other chronic disabling neurological diseases. For example, the annual cost of the routine treatment of multiple sclerosis in the era of disease modifying therapy exceeds 24 000€ per patient (24).
Although the surgical techniques associated with implantation of PNS devices for occipital nerve stimulation have improved, the complication rates are still high and refinements in both the technology and implantation techniques are required. In this study there were a total of 209 adverse events, 56 hardware-related AEs and 29 lead migrations. Of the device/ procedure related adverse events, 18 (8.6%) required hospitalization and 85 (40.7%) resulted in an additional surgery. Twenty-three of the 40 reported SAEs were classified as non-device or procedure-related events.
The 12-month follow-up is a strength of these data, but we do feel this may limit the ability to comment on several important issues. A follow-up period of at least 3 years would be ideal for determining the overall sustainability of the therapy as well as the cumulative AE profile and cost-benefit for patients with CM. However, an extensive clinical trial of this duration, including the retention and compliance of subjects with study protocol, visits and diary completion, already presents logistical challenges for patients and investigators. Our findings show sustained longer-term benefit from PNS in the occipital region for patients with CM. Together with the results from other controlled studies, we have identified reasonable endpoints for future studies. The number of headache days of moderate or severe intensity lasting for at least 4 hours is consistent with the definition of migraine, and measures the headaches that most disable patients. Responder rates (>30% and >50% reduction in headache days), headache-related disability, migraine-specific quality of life, and patient satisfaction are important endpoints to evaluate in this group of patients. These results, along with existing evidence demonstrating similar findings, suggest that it may be time for a pivotal registration phase III evaluating PNS in the occipital region for the preventive treatment of CM.
Clinical implications
Peripheral nerve stimulation in the occipital region is associated with sustained benefit over a 1-year period in some patients with chronic migraine, including some considered to have intractable disease. Patient-centered outcomes, such as quality of life, disability and patient satisfaction, are important measures in PNS device trials in patients with chronic migraine. Although the surgical techniques associated with implantation of PNS devices for occipital nerve stimulation have improved, the complication rates are still high and refinements in both the technology and implantation techniques are required.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
David W. Dodick M.D., within the past 12 months, has served on advisory boards and/or has consulted for Allergan, Amgen, Alder, Arteaus, Pfizer, Colucid, Merck, ENeura, NuPathe, Eli Lilly & Company, Autonomic Technologies, Ethicon J&J, Zogenix, Supernus, Labrys. Dr Dodick has received funding for travel, speaking, editorial activities or royalty payments from: IntraMed, SAGE Publishing, Sun Pharma, Allergan, Oxford University Press, American Academy of Neurology, West Virginia University Foundation; Canadian Headache Society; Healthlogix, Wiley, Universal Meeting Management, WebMD, UptoDate, Oregon Health Science Center, Starr Clinical, Decision Resources, Synergy. Because of Dr Dodick's affiliation with the journal's publisher, he was not involved in the editorial review process for this manuscript. Dr Mogilner received consulting fees, grant support and fellowship support from Medtronic Neurological, fees for serving on an advisory board, as well as grant support from St. Jude Medical and Boston Scientific. Dr Mogilner serves on editorial board of Neuromodulation. Dr Silberstein is on the advisory panel of and receives honoraria from Allergan, Artaeus, Electrocore, and Neuralieve. He serves as a consultant for and receives honoraria from Amgen, Labrys Biologics, MAP, and Zogenix. His employer receives research support from AGA, Allergan, Amgen, Cumberland, ElectroCore, Labrys, Merz, OptiNose, and Troy Healthcare. Dr Deer is a consultant for St. Jude Medical, Axionics, Medtronic, Nevro, Spinal Modulation and Bioness. Dr Sharan is a consultant for Medtronic, has received a speaker honorarium and research grant from St. Jude Medical, and is an owner of Integrate Care Pharmacy and ICVRx. Dr Slavin is a consultant and/or advisory board member for St. Jude Medical, Medtronic, Boston Scientific, Bioness, Greatbatch, Biotronik, W.L.Gore, and Nevro, receiving educational grants/research support from St. Jude Medical, Medtronic, Boston Scientific and Bioness, and honoraria from Karger and Wiley. Drs Mekhail, Vaisman, Reed, Trentman, Goldstein, Narouze and Ordia, report no conflicts of interest or specific financial disclosures.
Acknowledgements
First draft of this manuscript was prepared by the first author. All authors, including personnel from St Jude Medical, had the opportunity to provide input. All authors have provided approval of the final and submitted draft of this manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.