
Abstract
Background:
Chronic migraine (CM) is a debilitating neurological disorder with few treatment options. Peripheral nerve stimulation (PNS) of the occipital nerves is a potentially promising therapy for CM patients.
Methods:
In this randomized, controlled multicenter study, patients diagnosed with CM were implanted with a neurostimulation device near the occipital nerves and randomized 2:1 to active (n = 105) or sham (n = 52) stimulation. The primary endpoint was a difference in the percentage of responders (defined as patients that achieved a ≥50% reduction in mean daily visual analog scale scores) in each group at 12 weeks.
Results:
There was not a significant difference in the percentage of responders in the Active compared with the Control group (95% lower confidence bound (LCB) of −0.06; p = 0.55). However, there was a significant difference in the percentage of patients that achieved a 30% reduction (p = 0.01). Importantly, compared with sham-treated patients, there were also significant differences in reduction of number of headache days (Active Group = 6.1, baseline = 22.4; Control Group = 3.0, baseline = 20.1; p = 0.008), migraine-related disability (p = 0.001) and direct reports of pain relief (p = 0.001). The most common adverse event was persistent implant site pain.
Conclusion:
Although this study failed to meet its primary endpoint, this is the first large-scale study of PNS of the occipital nerves in CM patients that showed significant reductions in pain, headache days, and migraine-related disability. Additional controlled studies using endpoints that have recently been identified and accepted as clinically meaningful are warranted in this highly disabled patient population with a large unmet medical need.
Keywords
Introduction
Chronic migraine (CM) is a highly prevalent and disabling neurological disorder affecting approximately 2.0% of the general population (1,2). Chronic migraine is characterized by at least 15 headache days per month, of which at least 8 days meet diagnostic criteria for migraine without aura or respond to a migraine-specific acute medication (3). Compared with episodic migraine, individuals with CM experience significantly greater disability, economic burden, and impairments in health-related quality of life (4–7). Moreover, individuals with CM are up to four times more likely to suffer from major depression, and suicide attempts are more frequent than in the general population (8–10). There are few well-designed clinical trials evaluating preventive treatments for CM. Thus, there remains an urgent unmet medical need for effective, safe and evidence-based therapies for those who suffer from CM.
Peripheral nerve stimulation (PNS) in the occipital region has emerged as a promising treatment modality for a variety of medically refractory chronic primary headache disorders, including CM (11–20). The first published series of 25 CM patients treated with PNS showed an average headache day reduction of 38.1 out of 90 days from a baseline of 75.6 and an improvement of 88.7% in Migraine Disability Assessment (MIDAS), with minimal residual disability in 15/25 subjects who underwent implantation at an average follow-up of 18.3 months (12). Matharu et al. (13) reported significant improvement in six out of eight patients, whereas Schwedt et al. (15), showed significant improvements in headache frequency (improvement of 25 days from a baseline of 89/90 days), headache intensity (2.4 points from a baseline of 7.1 points), MIDAS scores (70 points from a baseline of 179 points), HIT-6 (11 points from a baseline of 71 points) and BDI-II scores (8 points from a baseline of 20 points) at a mean follow-up of 19 months. The Occipital Nerve Stimulation for the Treatment of Intractable Migraine (ONSTIM) study, the first published prospective, controlled feasibility trial evaluating the efficacy of occipital nerve stimulation (ONS) in CM, demonstrated at least a 50% reduction in headache frequency and/or a three-point intensity scale decrease in 39% of 66 patients treated with active PNS for 12 weeks (16). The PRecision Implantable Stimulator for Migraine (PRISM) study, which is the most recently conducted multicenter, double-blinded, randomized controlled study of ONS for the treatment of refractory migraine, showed a mean decrease of 5.5 migraine days/month in 63 patients who received active stimulation and a decrease of 3.9 days in 62 patients who received sham stimulation at 12 weeks. This difference was not statistically different. A sub-analysis to determine the impact of medication overuse showed a 5.0 headache day reduction in the active group and a 4.8 day reduction in the sham group in patients who were overusing medications, and a 5.9 day reduction in the active group and 2.6 day reduction in the sham group in patients not overusing acute headache medications. These results suggest that group differences may be more easily detected in patients not overusing medications.
The mechanism by which PNS may provide a reduction in attack frequency or pain severity is unclear, but may involve activation of central endogenous pain modulation networks (21). Clearly, further randomized controlled studies are necessary to determine the efficacy, safety, and tolerability of PNS in CM. The objective of this study was to evaluate the safety and efficacy of PNS in the management of pain and disability associated with CM. Results from the 12-week controlled phase of the study are presented here.
Methods
Sample size
A low response rate was expected in the Control group due to the requirement of a 50% reduction in mean daily average pain visual analog scale (VAS) scores. As a conservative estimate, this response rate has been assumed to be 15%. The population response rate among patients in the Active group was assumed to be 45%. Based on these estimates, 150 patients randomized 2:1 (100 in the Active group and 50 in the Control group) provide 80% power to reject the null hypothesis at the 5% level of significance.
Participants and study design
Key inclusion/exclusion criteria.

Surgical procedure
Only patients who underwent a successful trial (defined as at least 50% reduction in pain or adequate paresthesia coverage in the painful areas) to determine proper lead placement of the PNS device received implantation of the permanent system (n = 157). Patients who did not have a successful trial (n = 20) were classified as a screen failure and exited the study. For the permanent implant, the patients had leads (St. Jude Medical Neuromodulation) placed on either side of the midline caudally along the nerve or, more commonly, perpendicular to the course of the occipital nerves at the level of the craniocervical junction. Leads were placed either unilaterally or bilaterally depending on the pain distribution. A subcutaneous pocket was created for implantation of the implantable pulse generator (IPG (Genesis™; St. Jude Medical Neuromodulation)). A subcutaneous tunnel was made from the lead incision site to the pocket and the lead/extension was tunneled and connected to the IPG. The IPG was placed in the subcutaneous pocket and the incisions were closed.
Randomization and masking
After permanent implantation, patients were first stratified by use of alternative (including treatments such as acupuncture, herbal medications, and massage) or non-alternative therapies and then randomized into either an Active or Control group in a 2:1 ratio using a block size of three created by computerized software (SAS version 9.2). The 2:1 randomization was chosen to minimize the number of patients who received no stimulation, and to maximize the number of patients who received active stimulation for determination of device/procedure-related adverse events. Two sets of sealed envelopes were provided to each investigator and the appropriate set was opened by a sponsor representative. Both investigators and patients were blinded to treatment.
Programming
Patients in the Active group were programmed for appropriate stimulation. Patients in the Control group were given a sham programmer that did not communicate with the IPG. All patients were permitted to use their established pain medications and other treatment modalities (treatments already in use 8 weeks prior to baseline) at the same levels during the study, but new methods of pain control were prohibited.
Data collection
The primary outcome for this study was mean daily VAS measurements of average pain intensity recorded in a patient diary. A responder was defined as a patient with a reduction from baseline of 50% or greater together with no increase in average headache duration. Any patient who did not complete the 12-week controlled phase was treated as a non-responder. The primary endpoint of the study was a comparison of the proportion of responders in the Active group to those in the Control group at 12 weeks. A superiority delta of 10% was established for this study. Secondary outcomes included reduction in number of headache days (duration ≥4 hours with peak intensity reported as moderate or severe), MIDAS questionnaire, patient-reported headache pain relief (categorical and percentage) and adverse events. All variables were measured at 4 and 12 weeks post implant. If a patient completed less than 14 diary days during the assessment period, the patient’s value was set to missing for that period. The last observation carried forward (LOCF) approach was used for missing data. An intent-to-treat (ITT) analysis that included all patients (N = 157), as well as an analysis of only patients that met the criteria for intractable chronic migraine (ICM; N = 125), was performed for all variables at 12 weeks. The ICM population included patients who met the criteria for chronic migraine and had failed three or more preventative drugs, and were at least moderately disabled (MIDAS score of ≥11) at baseline.
Statistical methods
All statistical analyses were performed using SAS version 9.2. All statistical tests were two-sided with a significance level of 5%, unless otherwise specified.
The primary analysis was performed by placing a one-sided lower 95% confidence bound on the observed difference in proportions using a normal approximation. The results were presented with a one-sided 95% lower confidence bound (LCB) on the difference in proportions to compare with 0.10, along with a two-sided p value from a χ2 test for the test of superiority. A superiority margin of 10% was required by the FDA because of the additional risk imposed by the implantable device. A continuous proportion responder analysis of the primary outcome was also performed. In this analysis, a responder curve in which various definitions of a responder from a 10% reduction in mean VAS to a 100% reduction in mean VAS in increments of 10% (i.e. a 10% reduction, a 20% reduction, etc.) was generated. A χ2 test or a Fisher’s exact test (if a cell size was <5) was used to assess group differences.
The secondary analyses excluded patients who terminated prior to the end of the 12-week controlled phase. If a patient completed less than 14 diary days in a period, the patient’s value was set to missing for that period. Missing data were imputed using the LOCF approach as specified above. Reduction in the number of headache days (duration ≥4 hours with peak intensity reported as moderate or severe) from the baseline period to 12 weeks (normalized to 28 days) was analyzed using an analysis of covariance (ANCOVA) model with effects for treatment, study center, prior use of alternative therapy, and baseline number of headache days. A continuous proportion responder analysis was also performed for percent reduction of headache days. Migraine Disability Assessment scores were also analyzed using the ANCOVA model with effects for treatment, study center, prior use of alternative therapy, and baseline value. Categorical classification of headache pain relief was analyzed by the Cochran-Mantel-Haenszel procedure. Patient-reported percentage of headache relief was analyzed by an ANOVA model with the effect of treatment.
All randomized patients and all adverse events that occurred prior to the 12 week visit were included in the safety analyses. Differences between treatments in adverse event rates were tested using a χ2 test or Fishers exact test if cell size was <5.
Results
In this clinical trial, a total of 268 subjects were enrolled between 30 June 2005 and 20 August 2010 and 157 were implanted with a permanent system and randomized (Figure 1). Patient demographics and baseline characteristics are presented in Tables 2 and 3.
Enrollment. Patient demographics. Baseline characteristics.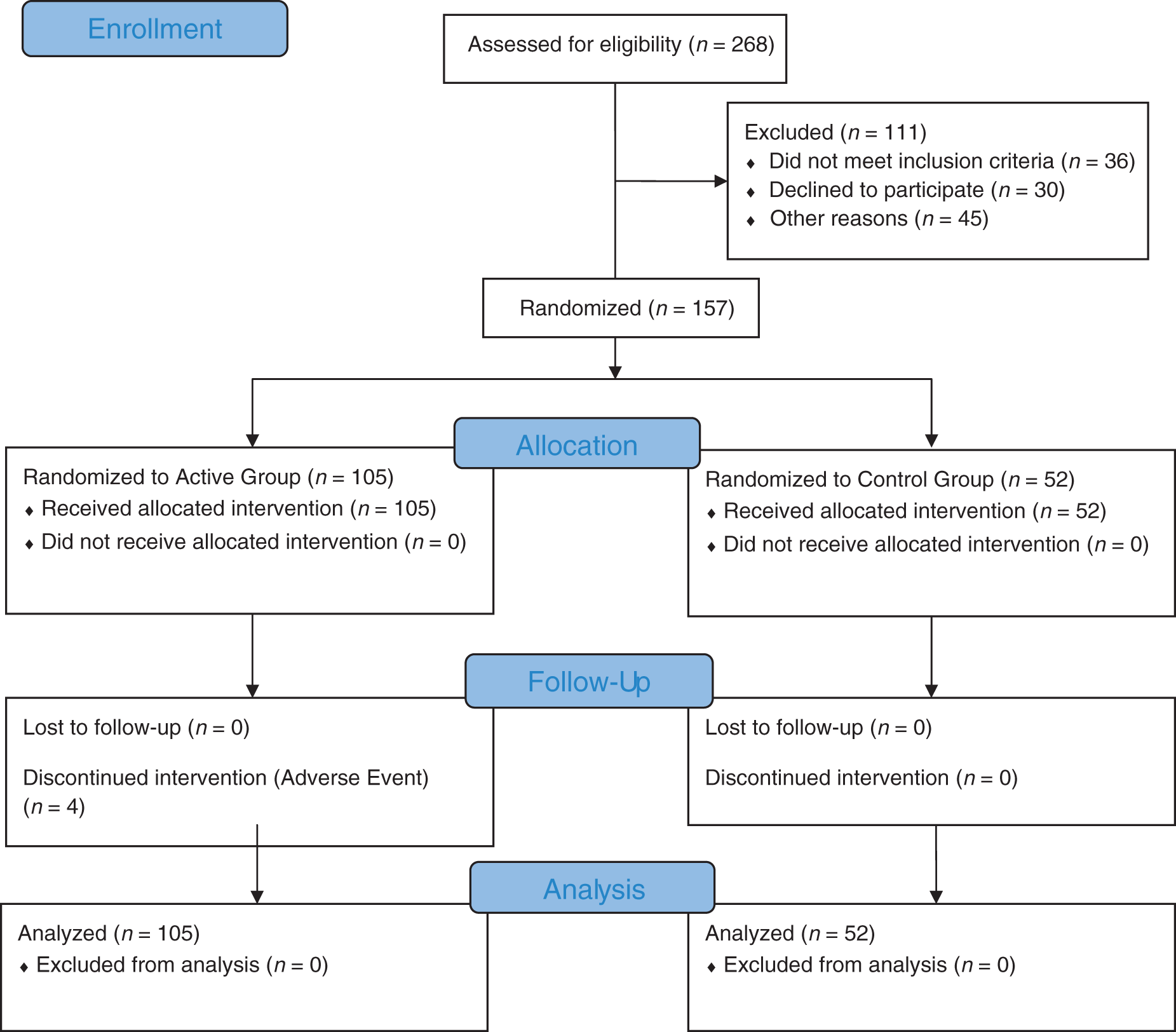


Primary outcome
For the ITT analysis, 18 patients (17.1%) in the Active group compared with seven patients (13.5%) in the Control group were classified as responders. This difference was not statistically significant (95% LCB of −0.06; p = 0.55). These results are shown in Figure 2. However, the continuous proportion responder analysis showed statistically significant group differences for the percentage of patients achieving a 10% (95% LCB of 0.12; p = 0.003), 20% (95% LCB of 0.09; p = 0.009), and 30% (95% LCB of 0.06; p = 0.02) reduction in headache pain. A continuous proportion responder analysis of the ICM population showed similar results with significant groups differences in the percentage of patients achieving a 10% (95% LCB of 0.19; p = 0.001), 20% (95% LCB of 0.13; p = 0.006), and 30% (95% LCB of 0.10; p = 0.011) reduction in headache pain. These results are displayed in Figure 3.
Percentage of responders based on 50% reduction from baseline in mean daily average pain intensity visual analog scale (VAS) measurements (primary outcome). The primary outcomes were analyzed for the ITT population only. In this population, 18 patients (17.1%) in the Active group compared with seven patients (13.5%) in the Control group were classified as responders. This difference was not statistically significant. Continuous proportion responder analysis based on mean daily average pain intensity visual analog scale (VAS) measurements for the intent-to-treat (ITT) (a) and intractable chronic migraine (ICM) (b) populations. This continuous proportion responder analysis was based on mean daily average pain intensity VAS measurements in patients with no increase in average headache frequency or duration. Statistically significant group differences were noted for the percentage of patients achieving a 10%, 20%, and 30% reduction in pain in both the ITT and ICM population.

Secondary outcomes
Reduction in headache days at 12 weeks is displayed in Figure 4. The difference between groups in the reduction of headache days was significant for both the ITT and ICM population (95% CI −5.4 to −0.8; p = 0.008 and 95% CI −7.0 to −1.7; p = 0.002, respectively). A continuous proportion responder analysis for percent reduction of headache days for the ITT population revealed significant group differences for the percentage of patients reporting a 10% (95% LCB of 0.06; p = 0.02), 20% (95% LCB of 0.07; p = 0.02), and 30% (95% LCB of 0.07; p = 0.02) reduction in headache days. Similar results were observed in the ICM population, with significant differences in the percentage of patients who achieved a 10% (95% LCB of 0.09; p = 0.013), 20% (95% LCB of 0.09; p = 0.015), 30% (95% LCB of 0.09; p = 0.014), and 40% (95% LCB of 0.06; p = 0.034) reduction in headache days. These results are shown in Figure 5.
Reduction in headache days from baseline to 12 weeks post implant for the intent-to-treat (ITT) (a) and intractable chronic migraine (ICM) (b) populations. For the ITT population, patients in the Control group reported a 14.9% reduction in the number of headache days and patients in the Active group reported a 27.2% reduction. For the ICM population, patients in the Control group reported an 11.4% reduction in the number of headache days and patients in the Active group reported a 28.3% reduction. The difference in the decrease in the number of headache days between groups was significant for both the ITT and ICM population. Continuous proportion responder analysis based on percent reduction in headache days from the baseline period to 12 weeks post implant for the intent-to-treat (ITT) (A) and intractable chronic migraine (ICM) (B) populations. Statistically significant group differences were noted for the percentage of patients achieving a 10%, 20%, and 30% reduction in the number of headache days in both the ITT and ICM populations. The percentage of patients achieving a 40% reduction in the Control group (13.5%) was also statistically lower than the percentage of patients reporting the same in the Active group (31.8%) in the ICM population.

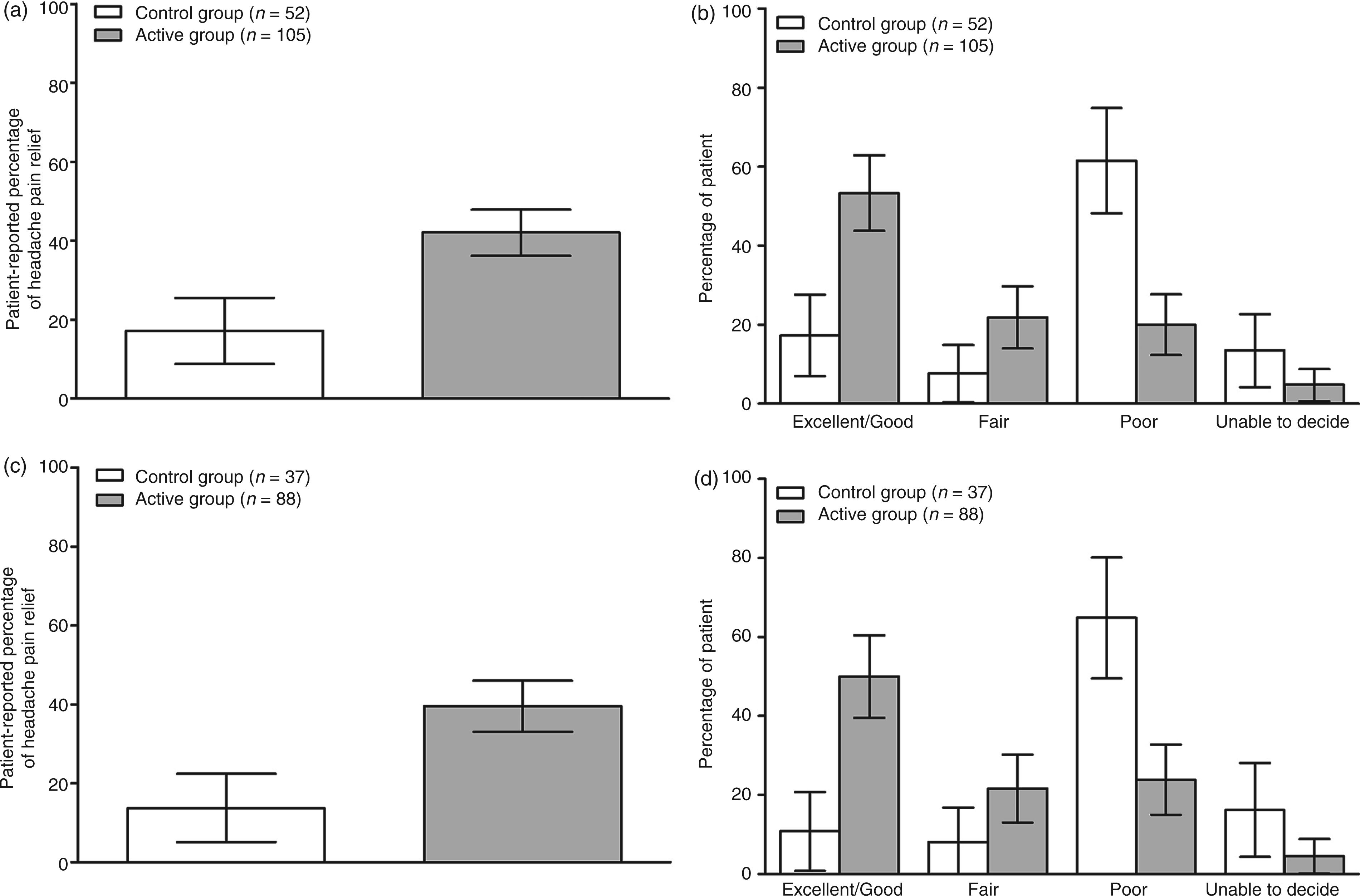
Migraine Disability Assessment scores at baseline and 12 weeks are shown in Figure 6. The MIDAS provides a grading system for disability as follows: Grade I (little or no disability; score of 0–5), Grade II (mild disability; score of 6–10), Grade III (moderate disability; score of 11–20), and Grade IV (severe disability; score of 21 or over). Migraine Disability Assessment scores were significantly reduced in the Active group compared to the Control group in both the ITT (95% CI −65.3 to −22.8; p = 0.001) and ICM (95% CI −71.2 to −20.2; p = 0.001) populations. Figure 7 shows categorical headache relief ratings and patient-reported percentage of headache pain relief. Significantly more patients in the Active group categorized their headache pain relief as “good” or “excellent” for both the ITT (p = 0.001) and ICM (p = 0.001) populations. Patients in the Active group also reported a significantly greater percentage of pain relief than those in the Control group for both the ITT (p = 0.001) and ICM (p = 0.001) populations.
Migraine Disability Assessment (MIDAS) scores at baseline and 12 weeks post implant for the intent-to-treat (ITT) (a) and intractable chronic migraine (ICM) (b) populations. In the ITT population, MIDAS scores for patients in the Control group were reduced by 20.4 points and scores for those in the Active group were reduced by 64.6 points. In the ICM population, MIDAS scores for patients in the Control group were reduced by 27.2 points and scores for those in the Active group were reduced by 72.9 points. These group differences were statistically significant for both the ITT and ICM populations. Patient-reported percentage of headache pain relief and categorical ratings of headache pain relief at 12 weeks post implant the for intent-to-treat (ITT) and intractable chronic migraine (ICM) populations. The difference in patient-reported percentage of headache pain relief between the Active and Control groups was statistically significant in both the ITT (a) and ICM (c) populations, as were the differences in the percentage of patients reporting excellent/good, fair and poor headache pain relief in both the ITT (b) and ICM (d) populations.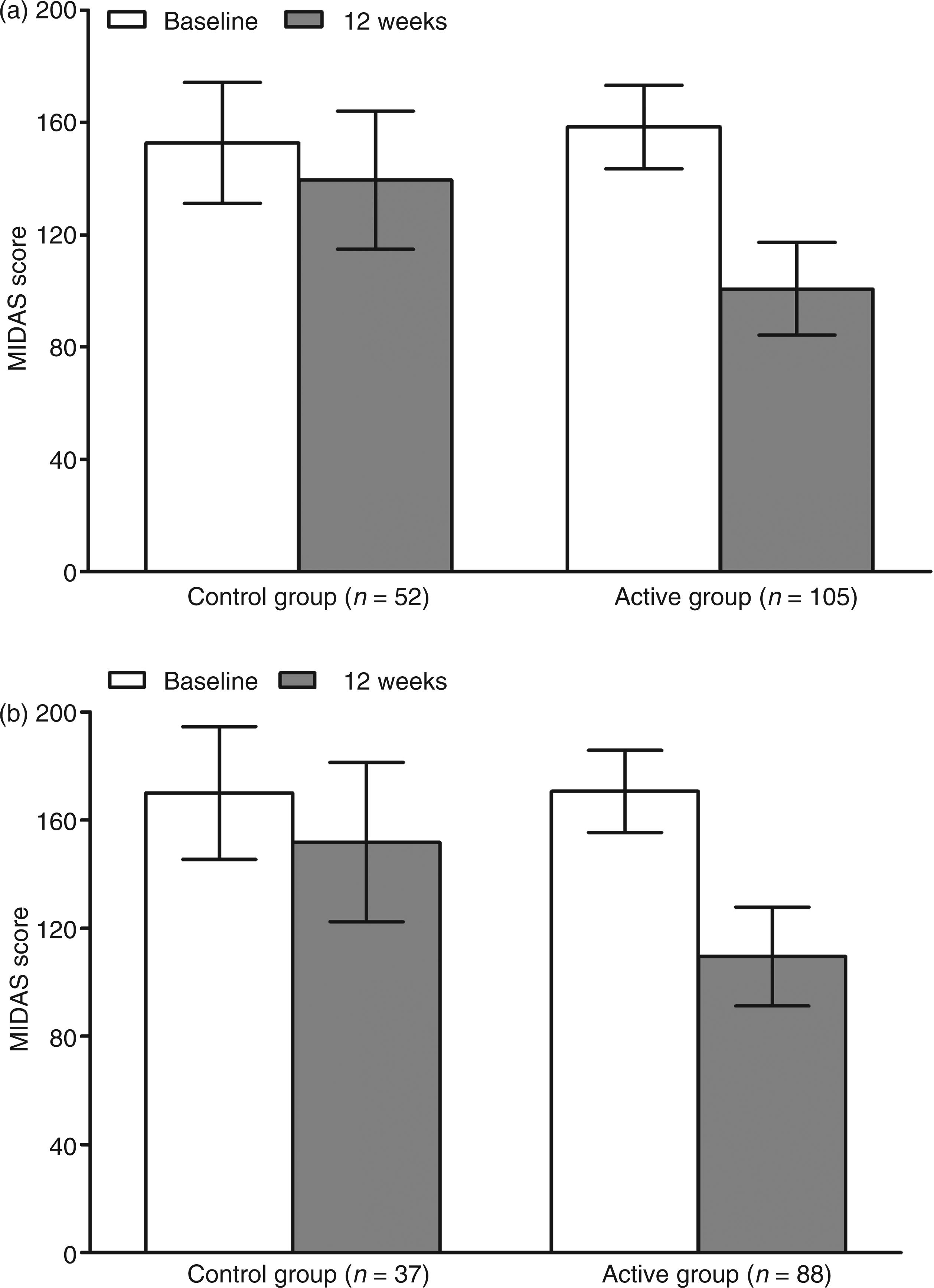
Adverse events
Summary of all adverse events during the 12-week controlled phase of the study.

Data are presented as the number and percentage of all adverse events. Overall, the most common hardware-related adverse event was lead migration which accounted for 18.7% of all events, the most common biological adverse event was persistent pain, and/or numbness at IPG/lead site which accounted for 21.5% of all events, and the most common stimulation-related adverse event was unintended stimulation effects which accounted for 6.5% of all events. Non-device/procedure-related events accounted for 9.4% of all events. There were no significant group differences for any of the adverse events.
Discussion
This randomized, double-blind, controlled clinical trial showed significant differences between the Active and Control groups for pain relief (30% reduction on VAS and patient-reported percentage), reduction in number of headache days, and MIDAS scores, but failed to show a significant difference in the number of patients who achieved a 50% reduction on the VAS for headache pain (primary endpoint). When this primary endpoint was established for the current study, very little was known about appropriate outcome measures in CM patients and even less was known about the impact of neurostimulation on the frequency, duration or severity of migraine headaches; it could either reduce pain during a headache, prevent headaches or both. Thus, pain reduction was largely chosen as the primary endpoint based on the history of neurostimulation in other pain conditions. At the time, the accepted standard for pain reduction was 50%; therefore, the endpoint was set at this level. Significant progress in the identification of appropriate outcome measures in CM patients, as well as an improved understanding of clinically meaningful changes in pain relief, have since been made (22). In 2008, recommendations put forth by the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) panel and the IHS established a 30% reduction in pain as clinically meaningful (22,23). Based on these recommendations, clinically meaningful pain relief (based on both VAS and direct patient reports) was achieved in the Active but not the Control group in both the ITT and ICM populations in the current study. However, limiting assessment of PNS of the occipital nerves in a disabling disorder such as CM to pain relief may fail to capture the potential benefit of the therapy as this may not necessarily be the most important objective. Furthermore, determining an average reduction in VAS score across a population of patients and comparing this to an average VAS score reduction in a control population may certainly hide individuals who derive significant benefit from the treatment.
The primary endpoint of more recent studies has been a reduction in headache days. Specifically, the ONSTIM feasibility study, which also assessed the effectiveness of occipital nerve stimulation, defined a responder as a subject who achieved a 50% or greater reduction in number of headache days per month or a three-point or greater reduction in average overall pain intensity compared with baseline (16). In the Phase 3 REsearch Evaluating Migraine Prophylaxis Therapy (PREEMPT) studies evaluating the efficacy of onabotulinumtoxinA for the prophylaxis of headaches in patients with CM, the reduction of the number of headache days was the primary endpoint (24,25) and based on the results of these pivotal studies, onabotulinumtoxinA was shown to be effective with clinically meaningful reductions in multiple other endpoints and was ultimately approved by the FDA for the treatment of CM. Prevention of headache days is intuitive, clinically relevant, and one of the primary reasons patients seek therapy. Therefore, assessments of CM treatments should not be limited to the degree of pain relief because this alone may not be necessary for clinically meaningful improvement, including reductions in headache-related disability. Assessments should also include improvement in mood, behavior, and migraine-specific quality of life and improvement in functional capacity as individuals with CM experience significant disability, economic burden, and impairments in health-related quality of life. The current study assessed a number of these dimensions in addition to pain relief and showed significant group differences for all in both the ITT and ICM populations.
The design of this study was unique from currently published literature due to randomization of enrolled subjects to either an Active group or a Control group, with the Control group receiving a “sham” device. The Control group was implanted with the PNS system and given the sham programmer that did not communicate with the IPG and therefore no stimulation was delivered. This group was then given a fully functional programmer after the assessments were completed at the 12 week visit. This unique study design was employed to protect the blind throughout the control period. However, it should be noted that some patients underwent a 3–5-day trial period prior to permanent implant of the device which provided them with experience with paresthesia. Whether experience with paresthesia during the trial period impacted the “blinding” during the controlled phase of the study remains unknown. Unfortunately, in the clinical practice of neurostimulation, there exists a need for a trial period in order to establish proper lead placement and determine whether the therapy will be tolerable and effective for a patient before subjecting them to an invasive surgical procedure. The ability to “trial” a patient prior to the permanent implant is considered a major benefit of neurostimulation and the inclusion of a trial period was considered important to the study design. Ultimately, it was determined that the benefits of having patients undergo a trial outweighed the risks of providing them with the experience of paresthesia, and the subsequent impact of this previous experience on blinding during the controlled phase of the study.
There were a total of 107 adverse events during the first 12 weeks of the study. Ten of these events were not related to the device or the procedure. Of the device/procedure-related adverse events, 46 (47.4%) were resolved with little to no risk to the patient (i.e. no action taken, reprogramming, or medication only), two (2.1%) required hospitalization, and the remaining 49 (50.5%) required an additional surgery. Although quite common, additional surgeries for peripheral (occipital) nerve stimulation systems are considered minimally invasive procedures because they do not involve entering the central nervous system or any body cavities. The surgeries are divided into several categories depending on the underlying problem and goals that the patient and physician are trying to achieve. A ‘revision’ procedure is a surgery that utilizes the existing implanted devices and moves them to a new location (i.e. moving IPG from one side of the body to another or moving a lead that has migrated). A ‘removal’ procedure is a surgery where existing implanted device(s) are removed from the patient. A ‘replacement’ procedure is a surgery where the existing implanted device(s) are removed from the patient and new devices are implanted. The majority of the additional surgeries involved revision, replacement, or removal of the lead(s). Although the number of lead migrations that occurred in the Active group was not statistically different from the number that occurred in the Control group, there were three times as many lead migrations in the Active group. This difference could have resulted because lead migration is not detected as often in the Control group because of the absence of stimulation. Lead migration is typically detected when a patient experiences a change in stimulation, which prompts an X-ray and subsequent determination of lead migration.
Additional information on adverse events will be reported in a subsequent publication.
Although the exact mechanism by which PNS exerts its effect in CM remains largely unknown, early experimental interest has centered on the trigeminocervical complex (TCC) and associated higher centers known to be related to the modulation of migraine headaches in particular, and pain modulation in general (13,26–29). The TCC is formed by the caudal trigeminal nucleus and portions of the upper three cervical dorsal horns (27). Nociceptive afferents from both the trigeminal nerve and the occipital nerves (C1-2-3) partially converge on the same second order neurons in the TCC and thus to a final common pathway to these higher centers (28). One study, which examined changes in regional cerebral blood flow measured by Positron Emission Tomography (PET) scans in eight CM patients who had marked benefit from use of ONS for 1.5 years, showed activation of some of these centers by ONS therapy (13). Specifically, various activation patterns were observed in the dorsal rostral pons, anterior cingulate cortex, pulvinar, and cuneus, which correlated with the stimulation-induced paresthesia and subsequent reduction in pain scores. Thalamic activation with PNS occurs without change in the underlying brainstem activation, suggesting a neuromodulatory mechanism for PNS therapy (29). A recent study in a preclinical model of migraine suggests that ONS may modulate nociceptive stimulation by increasing extracellular gamma-aminobutyric acid and blocking increases in glutamate (30). Studies like these and others aimed at determining the mechanism of action of stimulation of the occipital nerve will hopefully help to further refine the most appropriate patient population for this therapy.
Conclusions
There have been few well-designed clinical trials evaluating preventive treatments for CM, and only one interventional treatment, onabotulinumtoxinA, is currently approved by the FDA. Although the primary endpoint for this trial did not reach statistical significance, this is the first large-scale study of PNS for CM that showed significant and sustained reductions in pain, number of headache days, and migraine-related disability over the course of 1 year. These results were observed for both the ITT population and a subset of patients that meet the most current criteria for ICM, suggesting that patients that have exhausted all treatment options may benefit from PNS of the occipital region. Additional controlled studies using endpoints which have recently been identified and accepted as clinically meaningful are warranted to supplement these findings.
Footnotes
Funding
This work was supported by St. Jude Medical Neuromodulation Division.
Acknowledgements
Eugene Heyman, PhD provided statistical analysis as a paid consultant for St. Jude Medical Neuromodulation. Bing Zhang, PhD also conducted and verified all of the statistical analysis and all of the tabular results reported in this manuscript as a paid consultant of St. Jude Medical Neuromodulation. We thank all the investigators who contributed to the study and all the patients who agreed to participate in this study. Particularly, we thank our fellow investigators who were part of this clinical study who are not listed as authors: Stuart Black, MD, Herbert Markley, MD, Curtis Schreiber MD, Roger Cady, MD, Donald Bacon, MD, and James Banks, III, MD. We also thank all the research teams including the study coordinators, device programmers, and research nurses for all the time and support enrolling and assessing the patients and collecting all data for this study. Thanks to everyone who commented on a previous draft of the manuscript.
Conflict of interest
SS is on the advisory panel of and has received honoria from Allergan, Amgen, Capnia, Coherex, GlaxoSmithKline, Iroko Pharmaceuticals, Lilly, MAP Medtronic, Inc., Merck, Neuroalieve, NINDS, NuPathe, Pfizer and St. Jude Medical Neuromodulation. He is also on the speaker's bureau and has received honoraria from Allergen, Endo Pharmaceuticals, GlaxoSmithKline, Merck and Zogenix. He has consulted for and has received honoraria from Amgen, MAP, Nautilus, Novartis, OptiNose and Zogenix. DWD has consulted for Medtronic, Inc. and Boston Scientific. JS has consulted for Allergan, OMP, St. Jude Medical Neuromodulation, Autonomic Technologies, Neurocore, BMS and Neuroalieve and has received research grants from Allergan, Merck, St. Jude Medical Neuromodulation, Eli Lilly, Pfizer, Vanda, Forest Research Institute, Johnson and Johnson, Endo Pharmaceuticals, Astellas, BMS, SK Lifesciences, OptiNose and NuPathe. He is also on the speaker's bureau of Merck and OMP and owns stock in Pozen. BH has consulted for and has received honoraria from St. Jude Medical Neuromodulation. KVS has received honoraria, research support, educational grants and/or consulting payments from St. Jude Medical Neuromodulation, Medtronic, Inc., Boston Scientific, Bioness, Greatbatch, Integra, BSI, MESI, Karger and Elsevier. AS has consulted for and has received honoraria from St. Jude Medical Neuromodulation and Medtronic, Inc. He has also consulted for Zimmer Spine and is the founder of ICVRV. AM has received honoraria from St. Jude Medical Neuromodulation. TT has received research support from Medtronic, Inc. and has consulted for Boston Scientific Neuromodulation. TD is on the speaker’s bureau and advisory board of and has received honoraria from Bioness, MedaSys, St. Jude Neuromodulation and Spinal Modulation. He has also received research support from these companies. RL has consulted for St. Jude Medical Neuromodulation, Medtronic, Bioness and Spinal Modulation. RD is an employee of and owns stock in St. Jude Medical Neuromodulation. SW is an employee of St. Jude Medical Neuromodulation. There are no conflicts of interest for SN, KR, JV, JO, JG, NM and PW.
Contributors
All authors contributed to the recruitment of patients or performance of surgery. SS and DWD assisted in the interpretation of the outcome data and extensively revised the first and several additional drafts of the manuscript. SNW conducted the literature search and drafted the first version of the manuscript. All authors participated in the drafting of the manuscript and made substantial contributions to the final manuscript.