
Abstract
Background
Hemiplegic migraine (HM) and alternating hemiplegia of childhood (AHC) are rare episodic neurological brain disorders with partial clinical and genetic overlap. Recently, ATP1A3 mutations were shown to account for the majority of AHC patients. In addition, a mutation in the SLC2A1 gene was reported in a patient with atypical AHC. We therefore investigated whether mutations in these genes may also be involved in HM. Furthermore, we studied the role of SLC2A1 mutations in a small set of AHC patients without ATP1A3 mutations.
Methods
We screened 42 HM patients (21 familial and 21 sporadic patients) for ATP1A3 and SLC2A1 mutations. In addition, four typical AHC patients and one atypical patient with overlapping symptoms of both disorders were screened for SLC2A1 mutations.
Results
A pathogenic de novo SLC2A1 mutation (p.Gly18Arg) was found in the atypical patient with overlapping symptoms of AHC and hemiplegic migraine. No mutations were found in the HM and the other AHC patients.
Conclusion
Screening for a mutation in the SLC2A1 gene should be considered in patients with a complex phenotype with overlapping symptoms of hemiplegic migraine and AHC.
Keywords
Introduction
Hemiplegic migraine (HM) and alternating hemiplegia of childhood (AHC) are two rare episodic neurological brain disorders in which hemiplegia is a prominent symptom. Hemiplegic migraine is a subtype of migraine with aura that is characterized by transient hemiparesis during the aura phase (1). HM can present as a familial (familial hemiplegic migraine; FHM)) or a sporadic (sporadic hemiplegic migraine; SHM) disease. HM can occur in a pure form or with additional symptoms such as cerebellar ataxia, intellectual disability, and seizures (2). Mutations in the CACNA1A, ATP1A2 and SCN1A genes are well known to cause HM. The CACNA1A gene codes for a subunit of voltage-gated neuronal CaV2.1 calcium channels, the ATP1A2 gene for a subunit of Na+/K+ ATPases, and the SCN1A gene codes for a subunit of neuronal NaV1.1 sodium channels (3–5). Functional studies of HM gene mutations suggest a net increase in neurotransmitter levels (that is glutamate in the cortex) in the synaptic cleft as a key mechanism in HM (6), either due to an increased CaV2.1 function or reduced NaV1.1 channel function in excitatory and inhibitory neurons, respectively, or due to reduced functionality of glial Na+/K+ ATPases. Recently, truncating mutations in the PRRT2 gene have been reported in HM patients, of whom the majority also have phenotypes that are frequently associated with such PRRT2 mutations (i.e. paroxysmal kinesigenic dyskinesia, benign familial infantile seizures, and infantile convulsion choreoathetosis syndrome) (7–13). Based on this association, PRRT2 has been suggested as the fourth FHM gene.
AHC is characterized by intellectual disability and by recurrent attacks of hemiplegia, movement disorders, and seizures starting before the age of 18 months (14). The ATP1A3 gene was recently shown to be the major cause of AHC with mutations in more than 70% of patients (15–17). ATP1A3 codes for another subunit of Na+/K+ ATPases and is expressed in neurons of basal ganglia, cerebellum, and hippocampus. Na+/K+ ATPases are involved in the regulation of sodium and potassium gradients across glial (in the case of ATP1A2) or neuronal (in the case of ATP1A3) plasma membranes, thereby affecting sodium-coupled ion transport, neuronal excitability, and/or osmoregulation. In the respective cell types, FHM ATP1A2 and AHC ATP1A3 mutations result in a reduction of sodium-potassium pump activity (15–17).
In addition to typical AHC and HM patients there are also atypical patients who either do not fulfill all criteria of AHC or present with clinical symptoms of both AHC and HM. The atypical presentation makes it difficult, if not impossible, to determine the correct clinical diagnosis (18). Identification of gene mutations in atypical patients, therefore, may shed light on possible overlapping pathophysiological mechanisms between AHC and HM, and may confirm clinical diagnoses. Until now, an ATP1A2 mutation was identified in a Greek family with atypical AHC in which patients did not fulfill all diagnostic criteria of AHC (19,20). In addition, a CACNA1A mutation was identified in two German monozygous twins who displayed clinical features both of AHC and FHM (18), and a mutation was identified in the glucose transporter 1 GLUT1 gene SLC2A1 in an atypical AHC patient (21). The GLUT1 protein is pivotal for glucose transport into the brain, and mutations in SLC2A1 are a well-known cause of GLUT1 deficiency syndrome (22), which is characterized by developmental delay, medication-resistant epilepsy, and movement disorders including ataxia and dystonia. Because of the partially overlapping clinical manifestations of AHC and HM, the CACNA1A and ATP1A2 genes earlier were screened in typical AHC fulfilling all diagnostic criteria as well, but no mutations have been identified (23,24).
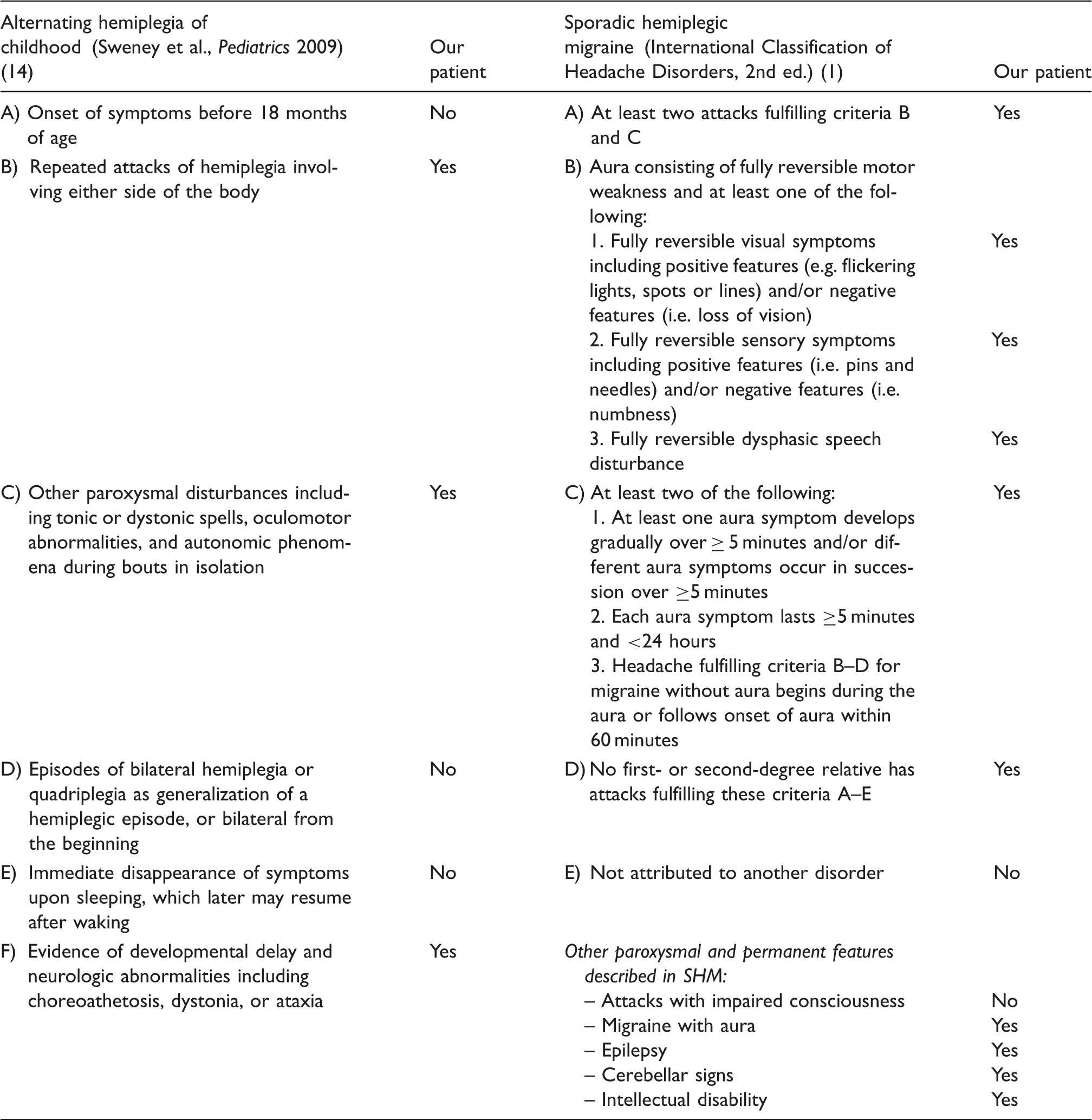
Diagnostic criteria for alternating hemiplegia of childhood and sporadic hemiplegic migraine (SHM).
Methods
Patients
For mutation screening of the SLC2A1 and ATP1A3 genes we selected 42 patients with HM (21 FHM and 21 SHM) for whom mutations in FHM genes (CACNA1A, ATP1A2, and SCN1A) had been excluded prior to the study by direct sequencing of all exons and flanking intronic regions. HM diagnoses were established according to the International Classification of Headache Disorders, 2nd ed. (ICHD-2; see Table 1 for SHM criteria) (1). We also included five ATP1A3-negative patients referred to us with a suspected AHC diagnosis in our screen of SLC2A1 mutations. Four of these patients fulfilled all diagnostic criteria for AHC, as outlined by Sweney et al. (see Table 1) (14). The fifth patient was atypical and met diagnostic criteria for SHM, but presented with a particularly severe phenotype of intellectual disability, seizures, and exercise-induced dystonia in addition to hemiplegic attacks, and thus symptoms fulfilled three of the six diagnostic criteria for AHC (see Table 1).
Three of the five AHC patients were sequenced for the CACNA1A and ATP1A2 genes, including the atypical patient. This was performed as part of a large mutation screen prior to the discovery of the ATP1A3 gene as a major gene for AHC. As these results were negative (23,24) we did not continue to screen for these genes in AHC patients. SCN1A screening was also performed in a group of AHC patients (data not published) and was negative as well. As SCN1A mutations have not been found in three large series of sporadic hemiplegic migraine patients with or without associated neurological symptoms (25–27), we did not perform SCN1A screening in the five AHC patients.
PRRT2 was screened only in the atypical AHC patient, as screening in a large subset of our HM patient cohort showed no mutations (data not published).
The local medical ethics committee approved the study and all patients or their parents provided written informed consent.
Mutation screening of ATP1A3 and SLC2A1
Ethylenediaminetetraacetic acid (EDTA) blood samples were collected from all patients and DNA was isolated using a standard salting out method (28). For the five AHC patients, genetic analysis of the ATP1A3 gene was performed by direct sequencing of all exons (and flanking intronic sequences) (ATP1A3 reference sequence: Genbank Accession Number NM_152296.4). For the 42 HM patients, ATP1A3 exons (and flanking intronic sequences) containing AHC-causing mutations (that is exons 5, 7–9, 13–16, 17, 18, 20–22) were sequenced. Genetic analysis of the SLC2A1 gene was performed by direct sequencing of all 10 exons and flanking intronic sequences (SLC2A1 reference sequence: Genbank Accession Number NM_006516). Multiplex ligation-dependent probe amplification was performed for SLC2A1 to search for intragenic deletions, using the SALSA multiplex ligation-dependent probe amplification (MLPA) kit P138 (MRC Holland, Amsterdam, the Netherlands) according to the manufacturer’s instructions.
Results
We did not identify any causal mutations in the ATP1A3 or SLC2A1 gene in the 42 HM patients and four typical AHC patients. In contrast, in the atypical patient with overlapping features of AHC and SHM, we identified a novel heterozygous p.Gly18Arg mutation (c.52G>C) in exon 2 of the SLC2A1 gene. This mutation was not found in 150 healthy controls nor in single-nucleotide polymorphism (SNP) databases (dbSNP, 1000G, Ensemble, ESP5400 and GoNL). It was also absent in the unaffected parents, and thus had occurred de novo. This patient showed a unique combination of symptoms, which were published 15 years ago without a genetic diagnosis (29). At the age of 20 years, she presented with progressive cerebellar ataxia and exercise-induced dystonia responding to oral corticosteroid treatment, and moderate intellectual disability. No ocular abnormalities were observed. From the age of 11 years, she had experienced episodes of hemiplegia, which did not resolve upon falling asleep. Hemiplegic attacks were always accompanied by inability to speak and confused speech. Flashing lights and ipsilateral numbness were present in some of the attacks. Hemiplegia, visual, and sensory symptoms were followed by contralateral headache and vomiting. Furthermore, she suffered from simple and complex partial seizures. Interictal electroencephalograms (EEGs) showed epileptiform activity, predominantly located right parietal. Brain magnetic resonance imaging scans (MRIs) were normal apart from a small non-specific white matter abnormality. Interictal cerebrospinal fluid (CSF) showed normal cell count, low glucose concentration of 2.4 mmol/l (reference: 2.5–3.7 mmol/l) and low lactate concentration of 1.2 mmol/l (reference: 1.3–1.9 mmol/l) (30). Blood glucose was not simultaneously measured.
Clinical follow-up at the age of 32 years revealed that no hemiplegic attacks had occurred during the last 10 years. Limb paresthesiae and hemianopsia lasting for several minutes without headache continued to occur and were classified as migraine with aura. Exercise-induced dystonia led to daily falls during walking. Ataxia had remained stable and seizures were never completely responsive to treatment with lamotrigine, sodium valproate, carbamazepine or clobazam. In retrospect, the CSF glucose and lactate concentrations were recognized as borderline low. The patient currently is considering starting a ketogenic diet.
Discussion
In this study we identified a novel pathogenic heterozygous SLC2A1 p.Gly18Arg mutation in an atypical sporadic patient with overlapping symptoms of both AHC and HM. In 2009, Rotstein et al. (21) found a p.Arg93Trp SLC2A1 mutation in a patient with atypical AHC features with intellectual disability, hemiplegic attacks (but otherwise not typical for HM either), episodes of ataxia, microcephaly and hypoglycorrhagia. Characteristically, mutations in SLC2A1 cause autosomal dominant glucose transporter type 1 (GLUT1) deficiency syndrome (31). The classical phenotype of GLUT1 deficiency syndrome consists of developmental delay, medication-resistant epilepsy, and movement disorders including ataxia and dystonia. A diagnosis of GLUT1 deficiency syndrome is suspected when hypoglycorrhachia (i.e. a low CSF glucose in a normoglycemic patient) is present. Symptoms improve with a ketogenic diet. Over the last few years it has become apparent that the clinical spectrum of the GLUT1 deficiency syndrome is broader (31,32).
Our patient expands the clinical phenotype of SLC2A1 mutations with episodes of HM and migraine with aura (without hemiplegia). The episodes of hemiplegia in our and Rotstein’s (21) patient differ from classic AHC because in contrast to classical AHC patients (i) the age of onset in these two patients was after 18 months; (ii) sleep had no beneficial effect on the attacks; (iii) dysconjugated eye movements were absent; and (iv) quadriplegic attacks were absent. Therefore, these patients do not meet the diagnostic criteria for AHC (Table 1) (1). Moreover, the hemiplegia in our patient meets the ICHD-II diagnostic criteria for SHM (Table 1) (1), but no mutations in CACNA1A and ATP1A2 were found, so far the only two genes with strong evidence for implication in the sporadic form of HM. The phenotype of our patient combines symptoms from GLUT1 deficiency syndrome and some of the severest presentations of SHM, both of which can present with intellectual disability, seizures and ataxia. The exercise-induced dystonia that is prominently present in our patient is not described in patients with SHM (2).
Different lines of evidence indicate that the p.Gly18Arg SLC2A1 mutation is the disease-causing mutation in our patient. The mutation had occurred de novo and was not found in healthy controls. Moreover, glycine at position 18 is a conserved amino acid in the first transmembrane segment of the GLUT1 protein. This segment is part of the central aqueous channel of the protein (33). Since the nonpolar glycine residue is replaced by a positively charged arginine residue, the mutation most likely interferes with normal GLUT1 function by disrupting glucose transport across the aqueous central channel. Finally, CSF glucose was low in combination with a low normal CSF lactate, which is within the range observed in SLC2A1 mutation carriers (30). The exercise-induced dystonia improved with oral corticosteroid treatment, possibly due to corticosteroid-induced hyperglycemia leading to increased GLUT1-mediated glucose transport in the brain, as has been shown by experiments in rats (34).
The absence of SLC2A1 mutations in the four classical AHC patients is in line with a previous mutation screening in 23 classical AHC patients (35) that was conducted before the identification of ATP1A3 as the causal gene in typical AHC patients (15,16). We did not identify causal mutations in ATP1A3 exons implicated in AHC in our HM patients. In conclusion, we propose that mutation screening of SLC2A1 should be considered in patients with a complex phenotype with overlapping symptoms of AHC and HM, especially if hypoglycorrhachia is detected in CSF. Diagnosing patients with a similar complex phenotype and SLC2A1 mutations may be of importance also because symptoms may ameliorate upon treatment with a ketogenic diet.
Clinical implications
Hemiplegic migraine and alternating hemiplegia of childhood (AHC), two neurological disorders with paroxysmal hemiplegia as a prominent part of their phenotype, have considerable clinical and genetic overlap. Mutations in the AHC genes ATP1A3 and SLC2A1 do not seem to play a role in patients with a typical hemiplegic migraine phenotype. Screening for a mutation in the SLC2A1 gene should be considered only in patients with overlapping symptoms of both disorders.
Footnotes
Funding
This work was supported by grants from the Netherlands Organization for Scientific Research (NWO) (903-52-291, M.D.F.; Vici 918.56.602, M.D.F.; Vidi 917-11-319, G.M.T), NWO “AGIKO-stipendium”; grant number 92003529, W.G.L.); the European Community (EC) (EUROHEAD, LSHM-CT-2004-504837, M.D.F.; EUROHEADPAIN (M.D.F. & A.M.J.M.v.d.M.) and the Centre for Medical Systems Biology (CMSB) in the framework of the Netherlands Genomics Initiative (NGI) (A.M.J.M.v.d.M).
Conflicts of interest
Dr Weller has nothing to declare. Dr Leen has received a grant for performing research on the subject of GLUT1 deficiency syndrome (NWO, ZonMW, “AGIKO-stipendium”; grant number 92003529). Drs Neville, Duncan, De Vries, Geilenkirchen, Haan and Kamsteeg have nothing to declare. Dr Ferrari reports grants and consultancy or industry support from Medtronic, Menarini, and Merck and independent support from NWO, ZonMW, National Institutes of Health (NIH), European Community, and the Dutch Heart Foundation. Dr Van den Maagdenberg reports independent support from NWO. Drs Willemsen and Scheffer have nothing to declare. Dr Terwindt reports grants and consultancy/industry support from Merck and Menarini, and independent support from NWO.