
Abstract
Background
LBR-101 is a fully humanized monoclonal antibody that binds to calcitonin gene-related peptide.
Objective
The objective of this article is to characterize the safety and tolerability of LBR-101 when administered intravenously to healthy volunteers, by presenting the pooled results of the Phase 1 program.
Methods
LBR-101 was administered to 94 subjects, while 45 received placebo. Doses ranged from 0.2 mg to 2000 mg given once (Day 1), as a single IV infusion, or up to 300 mg given twice (Day 1 and Day 14).
Results
Subjects receiving placebo reported an average of 1.3 treatment-emerging adverse events vs 1.4 per subject among those receiving any dose of LBR-101, and 1.6 in those receiving 1000 mg or higher. Treatment-related adverse events occurred in 21.2% of subjects receiving LBR-101, compared to 17.7% in those receiving placebo. LBR-101 was not associated with any clinically relevant patterns of change in vital signs, ECG parameters, or laboratory findings. The only serious adverse event consisted of “thoracic aortic aneurysm” in a participant later found to have an unreported history of Ehlers-Danlos syndrome.
Conclusion
Single IV doses of LBR-101 ranging from 0.2 mg up to 2000 mg and multiple IV doses up to 300 mg were well tolerated. Overt safety concerns have not emerged. A maximally tolerated dose has not been identified.
Introduction
Calcitonin gene-related peptide (CGRP) is a 37-amino acid neuropeptide derived from the gene encoding calcitonin by alternative splicing of microRNA (mRNA) and proteolytic processing of its precursor (1,2). CGRP occurs in two isoforms, α- and β-CGRP (3,4). α-CGRP is the predominant form in the peripheral nervous system, while the β-isoform is mainly present in the enteric nervous system (5). Radioimmunology studies demonstrate that CGRP is especially common in the trigeminal system, where up to 50% of the neurons produce it (6,7).
The potential role of CGRP in migraine pathophysiology was suggested more than 20 years ago (8,9) and since then our knowledge of the peptide and its role in the pathophysiology of migraine has increased substantially, leading to a robust interest in targeting CGRP to treat migraine. Clinical proof of efficacy for the acute treatment of migraine has been obtained with small-molecule CGRP receptor antagonists (10–12), but their development has been complicated by signs of liver toxicity associated with frequent use (13,14).
Monoclonal antibodies (mAbs) were first shown to have therapeutic activity in 1982 (15). In the past 30 years, the science of protein engineering and the clinical utility of mAbs have expanded dramatically. Currently there are more than 20 mAbs that are either humanized or fully human and are approved for human use (16). The utility of mAbs as therapeutics lies in part with their target-specificity, typically prolonged half-lives in humans, as well as reduced potential for hepatotoxicity and drug-drug interactions (17). Accordingly, CGRP mAbs offer an attractive opportunity for the preventive treatment of migraine if efficacy and safety can be demonstrated.
LBR-101 (formerly known as RN-307 or PF-04427429) is a fully humanized mAb that potently and selectively binds to both isoforms (α and β) of CGRP, blocking its binding to the CGRP receptors. LBR-101 is specific for CGRP and does not bind to the closely related family members amylin, calcitonin or adrenomedullin peptides. Two mutations were introduced into the constant region of the heavy chain to limit antibody effector functions (18,19). This loss of function prevents LBR-101 from stimulating antibody-dependent cell-mediated cytotoxicity and triggering complement-mediated lysis (20). The pharmacokinetic characteristics of mAbs make them attractive for the preventive treatment of headaches where CGRP plays a relevant role. The pharmacological properties of LBR-101 have been well characterized and the safety of the product has been extensively examined in the preclinical program.
Phase 1 data are inadequately disseminated. In a comprehensive review of the topic, it was found that only 17% of Phase I studies are published in scientific journals, and only a fraction of them are published in peer-reviewed journals targeting the audience that will ultimately make judgments about the value of the product (21). The findings led for calls by the World Health Organization and other bodies for publication of Phase I trial results in full (22). Accordingly, herein we characterize the safety and tolerability of LBR-101 when administered to healthy volunteers, by presenting the pooled safety and tolerability results from our entire Phase 1 program.
Methods
Across the Phase 1 program, five separate double-blind, placebo-controlled trials have been completed and LBR-101 has been administered intravenously (IV) to 94 healthy subjects, while 45 subjects received placebo. All studies were designed to study the safety and tolerability of the molecule. Being first in human, these studies were also used to characterize the pharmacokinetic properties of the medication.
Overview of study designs
Summary of clinical studies of LBR-101.

PK: pharmacokinetics; PD: pharmacodynamics; IV: intravenous.
Study B0141006 was distinct from the others since it also aimed to integrate pharmacodynamic readouts through measuring capsaicin flare inhibition up to one week after IV infusion of LBR-101. It is the only study in the Phase 1 program that used a cross-over design in a two-sequence, three-dosing period paradigm. For the purpose of this manuscript, we report on the adverse events (AEs) profile of the first dosed period only.
Study B0141007 tested multiple doses of LBR-101 at either 30 or 300 mg IV given two weeks apart, using a parallel design.
Each eligible subject was assigned a randomization sequence via an interactive Web-based system that contained the treatment assignment. The randomization schema was developed by the lead statistician.
Participants in all studies were generally healthy men and women (from 18 to 65 years of age); all participants signed informed consent forms. All studies were approved by investigation review boards (IRBs). Study 1 (approval number 09.11.0001) and study 8 (approval number 00003563) were conducted in the United States and approved by the “Independent IRB, Ft Lauderdale, FL,” which is now “Shulman Associates IRB Inc.” Studies 2, 6, and 7 were conducted in Belgium. They were reviewed and approved in parallel by the IRB at Université Libre de Bruxelles, Hôpital Erasme (approval OM 021, of June 29, 2010). The reported protocols and data are part of the regulatory package of LBR-101, and all IRB submissions and documentations follow the proper Food and Drug Administration (FDA) regulations.
Assessment of AEs
AEs were defined as any untoward medical occurrence in clinical study participants, with or without causal relationship to study drug. AEs observed after administration of the study drug or placebo were termed “treatment-emergent” AE (TEAEs) regardless of potential causality with the study drug. All subjects experiencing TEAEs were followed at appropriate time intervals until the event had resolved or until the event had stabilized and/or reached a new baseline.
All TEAEs were ranked as being mild, moderate, or severe by the primary investigator. Serious AEs (SAEs) were defined a priori as any untoward medical occurrence that at any dose resulted in death, was life threatening (i.e. the subject was at immediate risk of death at the time of the event), required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability/incapacity (i.e. a substantial disruption of the subject’s ability to carry out normal life functions), resulted in a congenital anomaly/birth defect, or any other medically important event, at the discretion of the investigator.
For all TEAEs, the reasonable possibility of its relationship with the study drug had to be assessed by the primary investigator. Treatment-related AE (TRAEs) were to be considered when one of the following situations was present: 1) a plausible temporal relationship between the onset of the AE and administration of the investigational product could be identified; 2) the AE could not be readily explained by the patient’s clinical state, intercurrent illness, or concomitant therapies; 3) the AE abated on discontinuation of the investigational product or dose reduction.
Vital signs, clinical laboratory tests and electrocardiogram (ECG)
Blood pressure, pulse rate and oral temperature were measured at screening, pre-dose, immediately after the end of the infusion and multiple times during the patients’ confinements in the clinics, as well as at all clinic visits.
Laboratory tests included serum chemistries, hematology, and urinalysis. Hematology, chemistry, coagulation, and urine safety laboratory tests were performed at multiple study times. The laboratory tests performed in this study, as well as criteria for clinically relevant abnormalities, are listed in Table 5 in the Appendix.
ECGs were recorded at screening, pre-dose on Day 1, immediately after the end of the infusion and five other times during the first day, as well as in all clinic visits. QTcF values were derived using Fridericia’s (QTcF) heart rate correction formula (23). Absolute values and changes from baseline for the ECG parameters QT interval, heart rate, QTcF interval, PR interval and QRS interval were assessed by cohort, treatment, and time post-dose.
In addition to the safety assessments described above, Protocol B014008 included complete ophthalmic assessments at baseline and at three time points after dosing (Day 28, Day 84, and Day 168).
Data analysis
Clinical data and vital signs were summarized using descriptive tables and summary statistics. Laboratory and other safety data were summarized as a function of any change (values outside of the reference range), as well as any clinical relevant changes, which were defined a priori.
Summary tables were stratified by dose and data were pooled across studies. In addition, comparisons for consolidated data for all LBR-101 exposures were contrasted with placebo. Placebo was also contrasted with LBR-101 doses of 100 mg and higher (100 mg, 300 mg, 1000 mg, 1500 mg, and 2000 mg), and with LBR-101 doses of 1000 mg and higher (1000 mg, 1500 mg, and 2000 mg).
Results
Demographics
A total of 94 subjects received at least one dose of LBR-101 while 45 received placebo. Among those receiving LBR-101, the mean age was 40.8 (standard deviation (SD) = 5.4) years, 69.1% were men and 88.2% were white. Among those receiving placebo, the mean age was 37.8 (6.1) years, 71.1% were men and 86.7% were white.
AEs
Adverse event (AE) profile across the Phase 1 dose range in subjects receiving different LBR-101 or placebo.

n: any event, treatment related or not. N: treatment-related event. Table 2 includes final data for the five Phase 1 studies.
TRAEs happened in 21.2% of subjects receiving LBR-101, compared to 17.7% in those receiving placebo. At doses of 100 mg of LBR-101 or higher, TRAEs happened in 22.4% of participants. At doses of 1000 mg or higher, they happened in 21.7% of participants (Figure 1).
Proportion of individuals with treatment-related adverse events across all studies.
Incidence of adverse events (AEs) as a function of dose.
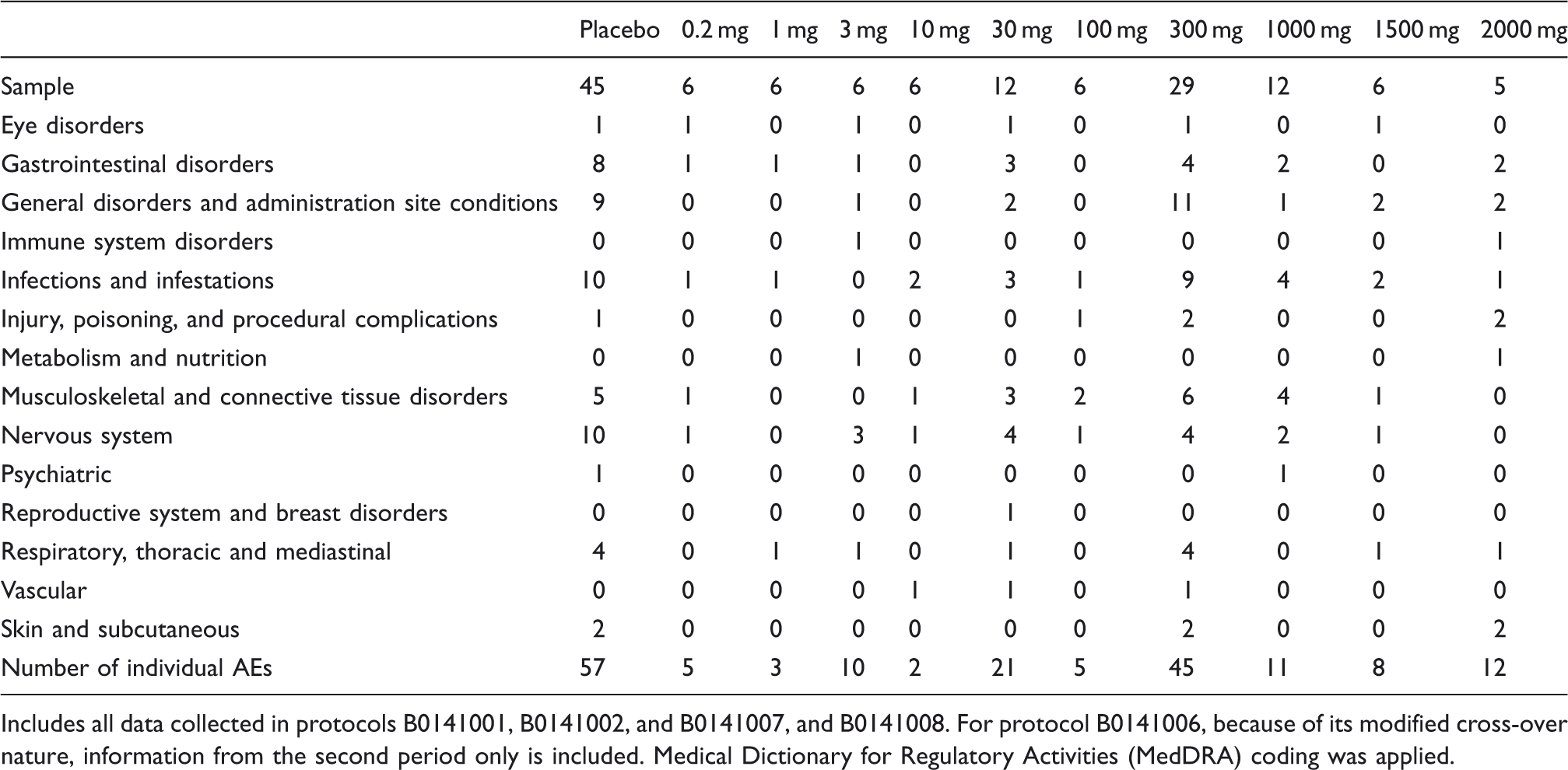
Includes all data collected in protocols B0141001, B0141002, and B0141007, and B0141008. For protocol B0141006, because of its modified cross-over nature, information from the second period only is included. Medical Dictionary for Regulatory Activities (MedDRA) coding was applied.
There have been no deaths. Two serious adverse events happened in individuals receiving LBR-101. The first happened in a participant receiving a single 300 mg dose of LBR-101, and consisted of a “thoracic aortic aneurysm aggravated.” The participant was later found to have an unreported history of Ehlers-Danlos syndrome. Causality was assigned at a time when the history of Ehlers-Danlos syndrome had not been identified. A second SAE, glaucoma, happened in a 64 year-old woman who received a single 1500 mg dose of LBR-101 and developed increased ocular pressure 84 days after exposure, justifying the assessment of noncausality.
One patient receiving placebo discontinued because of treatment-related adverse dermatitis, while one subject (2000 mg cohort) received a reduced dose because of a local injection site reaction.
Vital signs and laboratory findings
Blood pressure and laboratory abnormalities as a function treatment in participants of LBR-101 Phase 1 program.
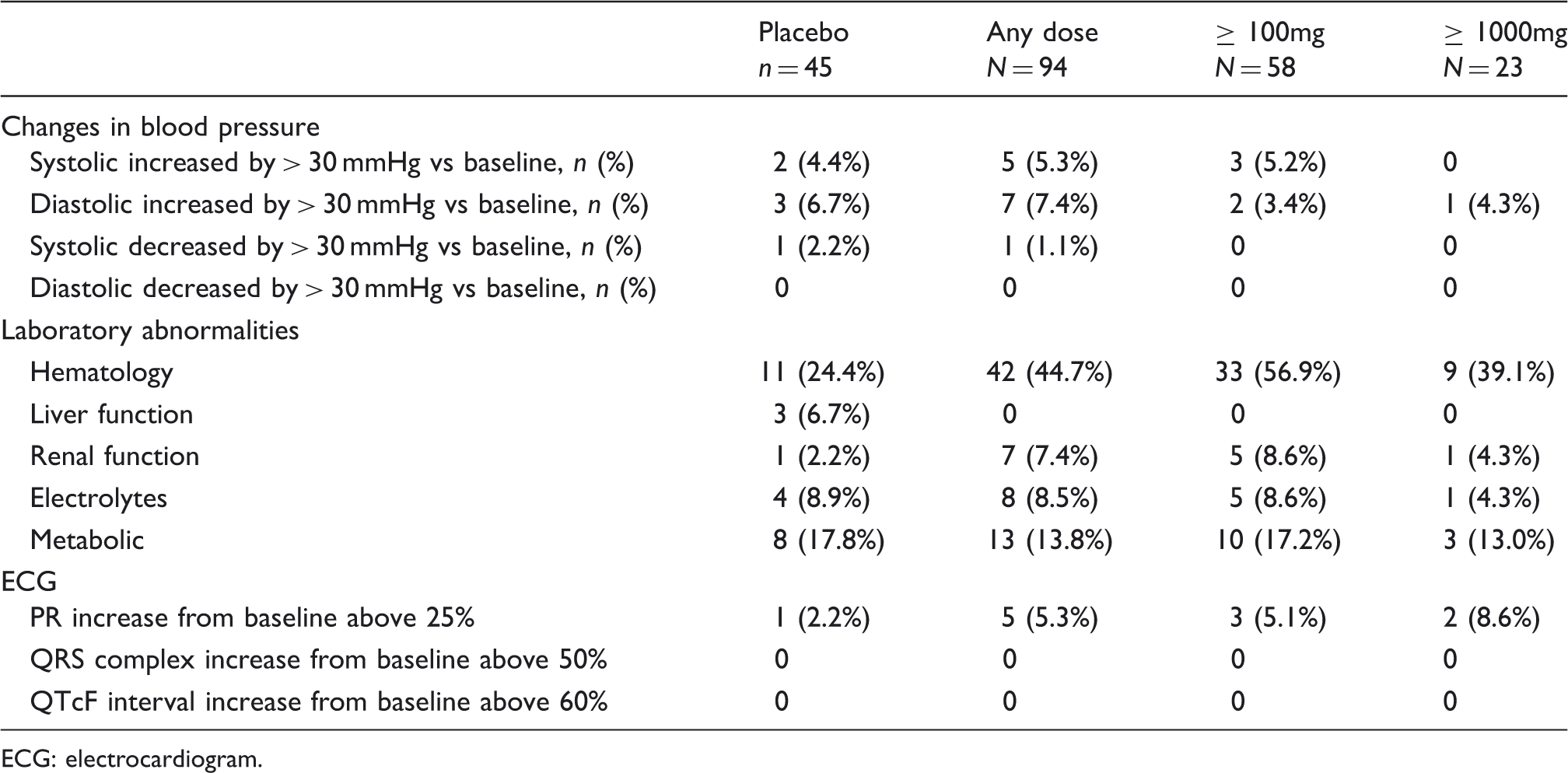
ECG: electrocardiogram.
Clinical laboratory findings were similar across placebo and LBR-101 (Table 4). In particular, liver function abnormalities, defined as any post-dose value outside the normal test range, were not observed for aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, alkaline phosphatase, or gamma-glutamyl transferase (GGT) among subjects receiving any of the studied doses of LBR-101. Limited effects on liver function tests were seen in three patients receiving placebo. All abnormalities described in Table 4 were mild in nature. We emphasize that Table 4 reports on any laboratory abnormality (defined as values outside of the normal range). Clinically significant abnormalities (as defined in the supplemental material) were not seen.
Discussion
CGRP is a well-studied neuropeptide that is relevant to migraine pathophysiology (7,24). At present, CGRP remains the most actively evaluated and probably best validated in development target in migraine drug research (25). CGRP receptor antagonists (CGRP-RA) disrupt the interaction of CGRP with its receptor and are being developed primarily for the acute treatment of migraine. Free CGRP and CGRP receptors can be targeted using mAbs, which are being developed for the preventive treatment of episodic and chronic migraine (CM) (13).
Five distinct, small-molecule CGRP-RA (the “gepants”) demonstrated clinical proof of efficacy for the acute treatment of migraine. Of the four, olcegepant (BIBN4096BS) was discontinued because of difficulties in developing an oral formulation (12); telcagepant (MK-0974) was discontinued because of concerns of liver toxicity after frequent use (13,14); and MK-3207, a molecule that was significantly more potent than telcagepant (26), was also discontinued because of concerns of liver toxicity. Positive findings were recently reported for a fifth molecule, BMS-927711 (27).
In addition to demonstrating proof of efficacy, the CGRP-RA clinical trials demonstrated the tolerability of this class with acute dosing and suggested that, as opposed to triptans, their use was not associated with vasoconstriction (28–30). Unfortunately, their potential use as preventive medications has been limited by issues of liver toxicity that, so far, has been the Achilles’ heel of these small-molecule agents in this class.
The rationale for developing mAbs that target CGRP is based on several assumptions that need to be validated during clinical development. The first assumption is that they are effective for the prevention of frequent migraines, which seems reasonable since the target (CGRP) has been validated. Nonetheless, efficacy of CGRP mAbs remains to be tested in Phase 2 and Phase 3 clinical studies. The second assumption, since mAbs do not cross the blood-brain barrier, is that they can deliver efficacy at peripheral sites of action (outside of the blood-brain barrier). CGRP-RA could theoretically exert their effects by blocking receptors for CGRP at several sites in the trigeminal and central nervous systems (31). Nonetheless, a study conducted using telcagepant found that the site of binding of the compounds in monkeys was mainly at the trigeminal ganglion that, at least in rodents, is located outside of the blood-brain barrier (32). The third assumption is that they have good tolerability, since poor tolerability is one of the most important barriers to adherence to preventive migraine care (33). The fourth assumption is that they are not limited by safety issues, including hepatotoxicity. Antibodies can be eliminated from the body by several pathways including binding to specific oligosaccharides in the liver, although the most frequent mechanism is via simple proteolytic catabolism, therefore bypassing the liver and decreasing the potential for liver toxicity. The clearance of LBR-101 happens with a half-life of around 45 days. Further, since mAbs are not substrates for, or inhibitors or inducers of the P450 cytochrome isoenzymes, drug-drug interactions by this mechanism are not expected (34,35).
The Phase 1 program provides data to partially support two of these assumptions in the context of short-term intervention in healthy volunteers. LBR-101 was found to be very tolerable across all doses. A maximally tolerated dose has not been identified, providing further support for the tolerability of this compound. Clinical laboratory safety samples were typically collected six times in the initial 24 hours after infusion, several other times doing the first week after infusion, and periodically for at least 90 days after administration. Hepatic function was within normal limits at all time-points in all subjects receiving LBR-101. This finding was expected since antibodies are not metabolized in the liver (36).
The studies reported herein investigated only one or two administrations of LBR-101, and future studies are needed to evaluate multiple doses of LBR-101 over longer time periods. It is also important to highlight that the demographics of our sample (mainly men) do not mimic the demographics of migraine. Because Phase 1 studies enroll healthy volunteers, they often do not focus on paralleling the demographics of the target disease unless there is good reason to think that gender influences the pharmacologic parameters of the drug. Phase 2 studies should, however, carefully mimic the demographics of the disease, not only focusing on efficacy, but also collecting data for pharmacokinetic analyses, in order to assess whether the disease itself, or disease-related demographics, influence these parameters. Furthermore, patients were healthy volunteers and assessments (e.g. vital signs and ECG) were conducted after subjects were resting quietly for at least 10 minutes in a supine position. The autonomic reflex (variation in blood pressure on rising from supine position) was not assessed.
The unmet medical need for the preventive treatment of episodic and CM is high (37). It is estimated that 35% of episodic migraineurs (and 100% of those with high frequency episodic migraine) should be offered migraine preventive therapies (38). However, many patients who qualify for preventive treatment do not receive it and they continue to have frequent attacks each month. Among those treated, some patients experience SAEs that preclude the continued use of therapies, while others receive no benefit (39). CM is less prevalent than episodic migraine, but because of the frequency of headaches and high degree of disability, all sufferers qualify for preventive therapy. Currently, only onabotulinumtoxinA has been approved for CM prevention (40). Accordingly, there is a clear need for the approval of additional preventive treatment options for CM. CGRP seems to play an important role in CM (41), and it has even been suggested to be a good marker for the disease (42).
LBR-101 is being developed for the preventive treatment of high frequency episodic migraine and CM. Single and multiple IV doses of LBR-101 ranging from 0.2 mg up to 2000 mg were well tolerated. Safety concerns have not emerged. No LBR-101-treated subject has experienced AEs leading to study discontinuation, or clinically relevant changes in vital signs, laboratory results, heart rate, and/or evidence of ECG effects associated with myocardial ischemia, infarction, conduction or repolarization abnormalities. A maximally tolerated dose has not been identified. These attributes support further clinical development of LBR-101 in patients with migraine.
Clinical implications
LBR-101 was found to be tolerable when given to normal volunteers at doses of up to 2000 mg. A maximally tolerated dose of LBR-101 was not identified. The safety profile of LBR-101, as assessed by the Phase 1 program, justifies moving the program to Phase 2.
Footnotes
Funding
This research was funded by the sponsors, Rinat, Pfizer, and Labrys Biologics.
Conflicts of interest
The authors are full-time employees of the sponsors.