
Abstract
Background
Understanding of the neuropathology leading to migraine pain has centered on either a vascular or neuronal origin. Sildenafil, a specific inhibitor of phosphodiesterase 5 (PDE5), induces migraine-like headache in a human headache model without concomitant artery dilation. The presence and activity of PDE3 and PDE5 is known in cerebral arteries. However, the presence in the neuronal part of the trigeminovascular pathway, i.e. the trigeminal ganglion and the possible co-localization with calcitonin gene-related peptide (CGRP), is not known.
Methods
Rat trigeminal ganglia were isolated and immunohistochemistry and in situ hybridization was applied. Evaluations of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) hydrolysis were performed using scintillation proximity assays.
Results
PDE3 and PDE5 were present and active in the trigeminal ganglia. A subset of PDE3- and PDE5-positive neurons contained CGRP. In contrast to cGMP, both sildenafil and cilostazol influenced cAMP hydrolysis.
Interpretation
Sildenafil may exert its effect on the neuronal part of the migraine pain pathway. In addition to the effects on cGMP signaling, sildenafil may indirectly affect cAMP signaling in the trigeminal ganglion. This result may suggest a common cAMP-related pathway for sildenafil, cilostazol, and CGRP in eliciting migraine pain.
Keywords
Introduction
In migraine pathophysiology, signaling molecules such as calcitonin gene-related peptide (CGRP), serotonin, and nitric oxide (NO) are involved in the efferent sensory signaling linked to the pain process (1–3). Although the signaling molecules associated with headache pain are known, what remains unknown is whether the origin of migraine pain is in peripheral mechanisms involving arterial blood vessels or in central mechanisms, e.g. trigeminal nerve signaling, the brain stem, and forebrain or perhaps a combination of both. Recent studies report a clear dissociation between dilation of arteries and alleviation of headache pain (4). Such findings are also in line with other results in which sildenafil induced a migraine-like headache without associated vasodilation (5,6). In addition, vasoactive intestinal peptide dilates cerebral arteries without concomitant induction of headache (7), and vasodilation by dipyridamole outlasts the headache response (8).
The CGRP neuropeptide is a key player in migraine (9) and activates adenylate cyclase by binding to G-protein-coupled membrane receptors, thus increasing intracellular cyclic adenosine monophosphate (cAMP) levels (10). The actions of NO occur through a parallel signaling pathway, increasing cyclic guanosine monophosphate (cGMP) production and activating the soluble guanylate cyclase (11). Migraine pain is alleviated by compounds that counteract CGRP and NO signaling, the CGRP antagonists and NO synthase inhibitors (12,13). Furthermore, migraine-like headache is induced by compounds potentiating cGMP and cAMP signaling, such as the phosphodiesterase (PDE) inhibitors sildenafil (6) and cilostazol (14), which inhibit PDE5 and PDE3, respectively. It is not, however, fully understood if the cAMP and cGMP signaling pathways interact via a relay point in the pain pathophysiology or if they represent two unassociated pain signaling pathways. A possible anatomical key relay point for either an interaction of the signaling pathways or the initiation or modulation of pain mechanisms could be the trigeminal ganglion or the trigeminal nucleus of the caudal brain stem.
CGRP and NO synthases localize to the trigeminal ganglion (15); however, the presence of PDE3 and PDE5, both modulators of the downstream cyclic nucleotide signaling, has not yet been studied in the trigeminal system.
The aim of this study, therefore, was to investigate the hypothesis that the cAMP-degrading PDE3 protein and the cGMP-degrading PDE5 protein are present in neurons of the rat trigeminal ganglion and that they co-localize with CGRP. Additionally, we sought to confirm that the PDEs are active and to investigate the effects of compounds associated with migraine pain signaling on cyclic nucleotide hydrolysis.
Materials and methods
Chemicals
Sildenafil was kindly provided by Peter Sandner, Bayer HealthCare, Wuppertal, Germany.
CGRP dimethyl sulfoxide (DMSO) and cilostazol were from Sigma-Aldrich, Broendby, Denmark.
Animals
The male Sprague-Dawley rats (Taconic Lille Skensved, Denmark) used in this study were 3 months old (300–350 g) and kept with free access to water and food on a 12:12 day:night cycle. The animals were acclimatized for one week in the animal facilities before inclusion in the experiments. All animal experiments were performed in accordance with the guidelines and regulations of the Danish Council of Animal Experiments (file: 2009/561-1664).
Immunohistochemistry
Perfusion fixation and cutting of sections
The rats were anesthetized by intraperitoneal injection of pentobarbital and immediately perfused through the left ventricle of the heart with heparinized (15,000 IU/l) phosphate-buffered saline (PBS) (pH 7.4) for three minutes followed by fixation with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. The rats were euthanized from noon to late afternoon. The trigeminal ganglia were removed and post-fixed in 4% paraformaldehyde overnight at 4℃. The ganglia were transferred to 20% sucrose for 24 hours and then frozen in Tissue-Tek® O.C.T.™ on dry ice and stored at –80℃ until sectioning. The tissues were sliced in a cryostat into sections 12–16 µm thick (Leica, Herlev, Denmark) and subsequently placed on Menzel SuperFrost slides (Thermo Fischer Scientific, Copenhagen, Denmark).
Fluorescent histochemistry
List of antibodies applied in the experiments.

PDE: phosphodiesterase; IHC: immunohistochemistry; WB: western blot.
Peroxidase immunohistochemistry
Methods for peroxidase immunohistochemistry were similar to the above, differing after the second application of ABC-Vectastain: The sections were washed in 0.05 M Tris-HCl buffer, pH 7.6, and incubated for five to 15 minutes at room temperature with a solution of 0.05% diaminobenzidine and 0.01% H2O2 in Tris-HCl buffer, pH 7.6. After a washing in distilled water, the sections were mounted, dehydrated, and cover-slipped with Pertex®, and photographed with a Zeiss AxioCam HR CCD camera.
Validation of antibodies by Western blotting
To validate the antibodies used for immunohistochemical studies, Western blot was performed. The rats were anesthetized with carbon dioxide and subsequently decapitated. The trigeminal ganglia were excised and immediately homogenized in lysis buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 0.5% Triton X-100), pH 7.4, containing a Complete® tablet (Roche, Hvidovre, Denmark) and PhosSTOP (Roche, Hvidovre, Denmark).
The cleared lysates were adjusted to 20 µg protein per sample, and the Western blot was performed as described in Schankin et al. (16) and run using 4%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (Kem-en-tec, Taastrup, Denmark). The proteins were transferred to a nitrocellulose membrane (Hybond ECL, GE Healthcare, Brøndby, Denmark) using wet electroblotting. The membranes were blocked for one hour in 2% ECL Advanced Blocking Agent (Hybond ECL, GE Healthcare, Brøndby, Denmark), incubated with the relevant primary antibody overnight at 4℃, washed in TBS-T (Tris-buffered saline with 0.01% Tween-20) and subsequently incubated with secondary antibody for one hour at room temperature. Membranes were developed using ECL Advanced Western Blot Detection Kit (GE Healthcare, Brøndby, Denmark) and imaged using an LAS-4000 digital imaging unit (GE Healthcare, Brøndby, Denmark).The antibody concentrations are listed in Table 1.
Radiochemical in situ hybridization
List of oligo mRNA probes used for in situ hybridization.

PDE: phosphodiesterase; mRNA: microRNA. For PDE3B hybridization, a 1:1 mixture of oligos 1 and 2 was used.
The in situ hybridization was performed as described by Larsen et al. (17). By use of a terminal transferase, the probes were end-labeled with S35-dATP (Perkin Elmer, Skovlunde, Denmark) by mixing 10 pmol of probe with 10 µl reaction buffer (Roche Mannheim, Germany), 24 ul diethylpyrocarbonate (DPEC)-treated water, 25 mM CoCl2, 5 µl S35-dATP, and 1 µl recombinant terminal transferase (Roche Mannheim, Denmark) for 75 minutes at 37℃. The reaction was stopped by adding 5 µl 0.2 M ethylenediaminetetraacetic acid (EDTA) (pH 8), and the probe was purified using an IllustraProbeQuant™ G-50 micro column (GE Healthcare, Denmark) according to the manufacturer’s instructions.
The sectioned tissue was washed twice in PBS and acetylated in 0.25% acetic anhydride in 0.1 M triethanolamine/0.9% NaCl for 10 minutes. The slides were then dehydrated in increasing percentages of ethanol (70%, 80%, 95%, and 100%) followed by delipidation in 100% chloroform for five minutes and then partial rehydration in 100% ethanol and 95% ethanol and before finally being left to dry.
For hybridization of the sections, the probes were diluted in hybridization buffer containing 50% v/v formamide, 4× saline-sodium citrate (SSC) solution (150 mM NaCl, 15 mM sodium citrate, pH 7), 1× Denhardt's solution (0.02% BSA, 0.02% polyvinylpyrrolidone, 0.02% Ficoll), 10% (w/v) dextran sulfate, 10 mM DTT, 0.5 mg/ml salmon sperm DNA, and 0.5 mg/ml yeast tRNA). The sections were incubated in a humid incubator overnight at 37℃. The following day, the sections were washed in 1× SSC solution for 4 × 15 minutes at 55℃ and 2 × 30 minutes at room temperature, rinsed in deionized water, and dried under warm air. Sections were then exposed to X-ray film for 14 days at 4℃ and developed. The developed X-ray film was captured by a high-performance black and white CCD-camera (Cohu, San Diego, CA, USA) equipped with a macro lens (Computar, Tokyo, Japan) by use of the software ScionImage 1.42 (Wayne Rasband, NIH, Bethesda, MD, USA)
The hybridized sections were subsequently dipped in a photographic emulsion diluted with water 1:1 (Amersham, Hillerød, Denmark) at 40℃ and exposed for four weeks. The sections were then developed for five minutes in a solution containing 0.45% amidol (Merck, Hellerup, Denmark), 0.08% potassium bromide (Merck, Hellerup, Denmark), and 1.8% sodium sulfite (Merck, Hellerup, Denmark), then washed in distilled water, fixed for 10 minutes in 70% ethanol, and counterstained for one minute in cresyl violet solution (0.1% cresyl violet in 1% acetic acid). Sections were viewed and photographed using a Zeiss Axiophot microscope.
Activity assay
Trigeminal ganglia were excised after opening the skull of each decapitated Sprague-Dawley rat. The ganglia were subsequently homogenized in PBS pH 7.4 containing Complete® protease inhibitor (Roche, Hvidovre, Denmark) and PhoSTOP® (Roche, Hvidovre, Denmark) by sonication. The samples were centrifuged 20 minutes at 14,000 rpm at 4℃ and kept on ice until initiation of the assay. The activity assay was performed using the Phosphodiesterase [3H] cGMP SPA Enzyme Assay or Phosphodiesterase [3H] cAMP SPA Enzyme Assay from GE Life Sciences (Skovlunde, Denmark) according to the manufacturer’s instructions. The assay was carried out in the presence of either 1 mM ethylene glycol tetraacetic acid (EGTA) or 4 µg/ml calmodulin and 200 µM CaCl2 with different concentrations of either sildenafil, cilostazol, or CGRP to a total assay volume of 90 µl. Prior to assay start, each sample was pre-incubated with either vehicle (DMSO or water) or treatment (sildenafil, cilostazol, or CGRP) for 10 minutes at room temperature. The assay was initiated by addition of either 10 µl of radioactive [3H]cAMP or [3H]cGMP and incubated for 15 minutes at room temperature. The assay was terminated by adding 50 µl of Yttrium SPA beads dissolved in 18 mM ZnSO4. The activity of each sample was determined using a MicrBeta2 microplate counter (Perkin Elmer, Skovlunde, Denmark). All conditions were run in triplicate and experiments were repeated five times.
Experimental design
The effect of sildenafil and cilostazol both on cAMP and cGMP hydrolysis was investigated using three treatment concentrations. Cilostazol was used at 0.1, 1, and 10 µM whereas sildenafil was used at 0.01, 0.1, and 1 µM, with each dissolved in DMSO. Further, in the case of sildenafil, because it also has an affinity for phosphodiesterase 1 (PDE1 IC50 = 281 nM), the PDE1 activators calcium and calmodulin (Ca/CaM) were added to some samples to determine the amount of hydrolytic activity originating from PDE1.
The effect of CGRP on cyclic nucleotide hydrolysis was investigated to elucidate a possible interaction of CGRP in influencing PDE activity. For this purpose, a single dose of 0.01 µM of CGRP was used.
Data and statistical data
Results are reported as mean ± SEM and were compared using Student’s t-tests or one-way analysis of variance (ANOVA). The differences were considered significant if p < 0.05. Statistical tests were performed using GraphPad Prism® software.
Results
Localization of PDE5, PDE3, and CGRP in rat trigeminal ganglion
Immunohistochemistry
The localization of PDE3 and PDE5 in the rat trigeminal ganglion was performed immunohistochemically by enzyme labeling of antibodies and by fluorescent detection. The majority of neurons in the ganglion showed positive staining for PDE3A, PDE3B, and PDE5A (Figures 1 and 2). The immunoreactivity was located in the cytoplasm of the perikarya and extended out into the processes emerging from the perikarya. The immunoreactive neurons were not confined in a specific area. In the peroxidase-stained sections, the immunoreactivity had a granular appearance in the cytoplasm, which was especially prominent for PDE3B (Figure 1(b)).
Immunohistochemical analysis of PDE3A, 3B, and 5A in the rat trigeminal ganglion. (a) PDE3A-immunoreactive neuronal cell bodies. Staining is granular (indicated by arrows). (b) PDE3B-immunoreactive neuronal cell bodies. Staining appears granular (indicated by arrows). (c) PDE5A-immunoreactive neuronal cell bodies (indicated by arrows). Bar = 50 µm. Immunohistochemical double-staining of rat trigeminal ganglion. (a) PDE3A-immunoreactive neuronal cell bodies (green). (b) PDE3B-immunoreactive neuronal cell bodies (green). (c) PDE5A-immunoreactive neuronal cell bodies (green). (d–f) CGRP-immunoreactive neuronal cell bodies (red). (g) Merged image of PDE3A-immunoreactive (green) and CGRP-immunoreactive (red). (h) Merged image of PDE3B-immunoreactive (green) and CGRP-immunoreactive (red) neuronal cell bodies. (i) Merged image of PDE5A-immunoreactive (green) and CGRP-immunoreactive (red) neuronal cell bodies. Yellow indicates co-localization.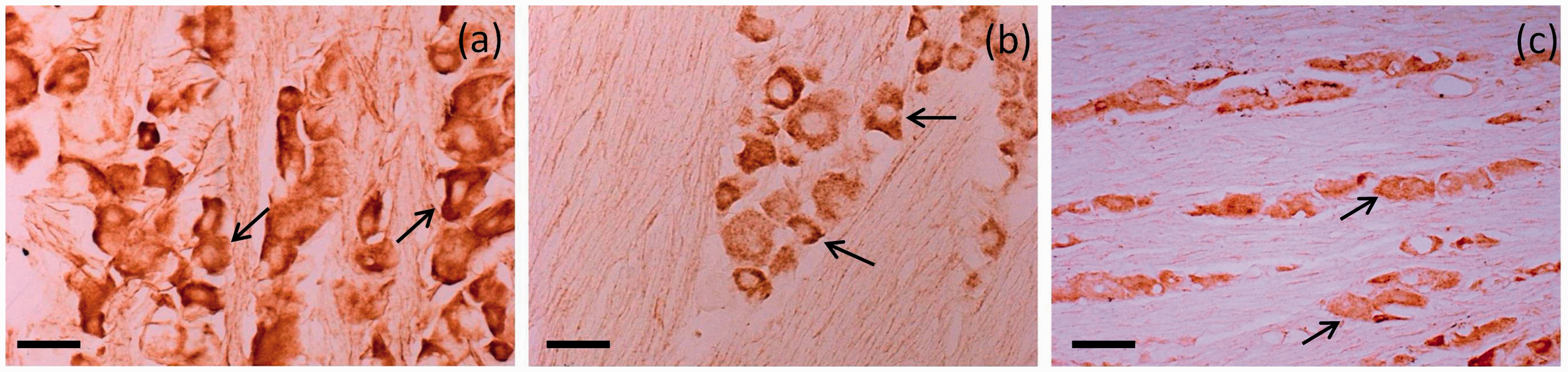

To investigate possible co-localization of PDE3 and PDE5 with CGRP, fluorescent detection was employed. As seen in Figure 2(a) to (c), most neurons were positive for PDE3A, PDE3B, and PDE5A. The CGRP staining was less frequent and present in approximately 15%–20% of neurons in the ganglion (Figure 2(d) to (e)). As shown in Figure 2(g) to (i), the enzymes PDE3A, PDE3B, and PDE5A were co-localized with CGRP in about 20%–30% of the trigeminal neurons. The co-localizing neurons were evenly distributed in the ganglion and not confined to any specific region.
To further validate the antibodies, Western blot was performed confirming that the antibodies recognize proteins of the expected sizes: PDE3A 110 kDa, PDE3B 135 kDa and PDE5A 95 kDa (data not shown).
In situ hybridization
By use of radiochemical in situ hybridization, a strong hybridization signal was obtained on the X-ray film with antisense probes detecting mRNA-encoding PDE3B and PDE5A (Figure 3(b) and (c)). A signal for PDE3A (Figure 3(a)) was also present in the trigeminal ganglion but of lower intensity compared to PDE3B and PDE5A. The hybridization of the sections with the sense probe did not result in a signal above background level (Figure 3(d)). The PDE signals were confined to the area of the trigeminal ganglion containing the sensory neurons. Nerve fibers extending both peripherally and centrally from the ganglion did not bind to the probe (Figure 3(a) to (c)). The highest signal was confined to cellular structures in the ganglion, with a less intensive signal between the cells. When the hybridized sections were dipped in the photographic emulsion and developed, the signal was clearly localized to the cytoplasm of the neuronal cell bodies of the ganglion (Figure 4).
In situ hybridization for PDE3A, PDE3B and PDE5A. Montage of autoradiographs of rat trigeminal ganglia hybridized for mRNA-encoding PDE3A, PDE3B, and PDE5A with S35-labeled antisense oligoprobes. (a) The PDE3A signal is of low intensity, but a clear delineation is seen between the nerve and the ganglion (black arrow). (b) The PDE3B hybridization signal exhibits strong intensity in the trigeminal ganglion (black arrow). (c) The PDE5A hybridization signal is strong, and individual neurons can be seen labeled in the ganglion (white arrow). (d) Results of a sense control hybridization using the reverse complement to the probe used in Figure 3(b). In situ hybridization for PDE5A (dipped section). Photomicrograph of part of a trigeminal ganglion of the rat, which has been hybridized for mRNA-encoding PDE5A. After hybridization, the section was dipped in a photographic emulsion and developed after three weeks of exposure. After development, the section was counterstained with 0.5% cresyl violet. The grains are located above nearly all the neurons (arrows). Bar = 100 µm.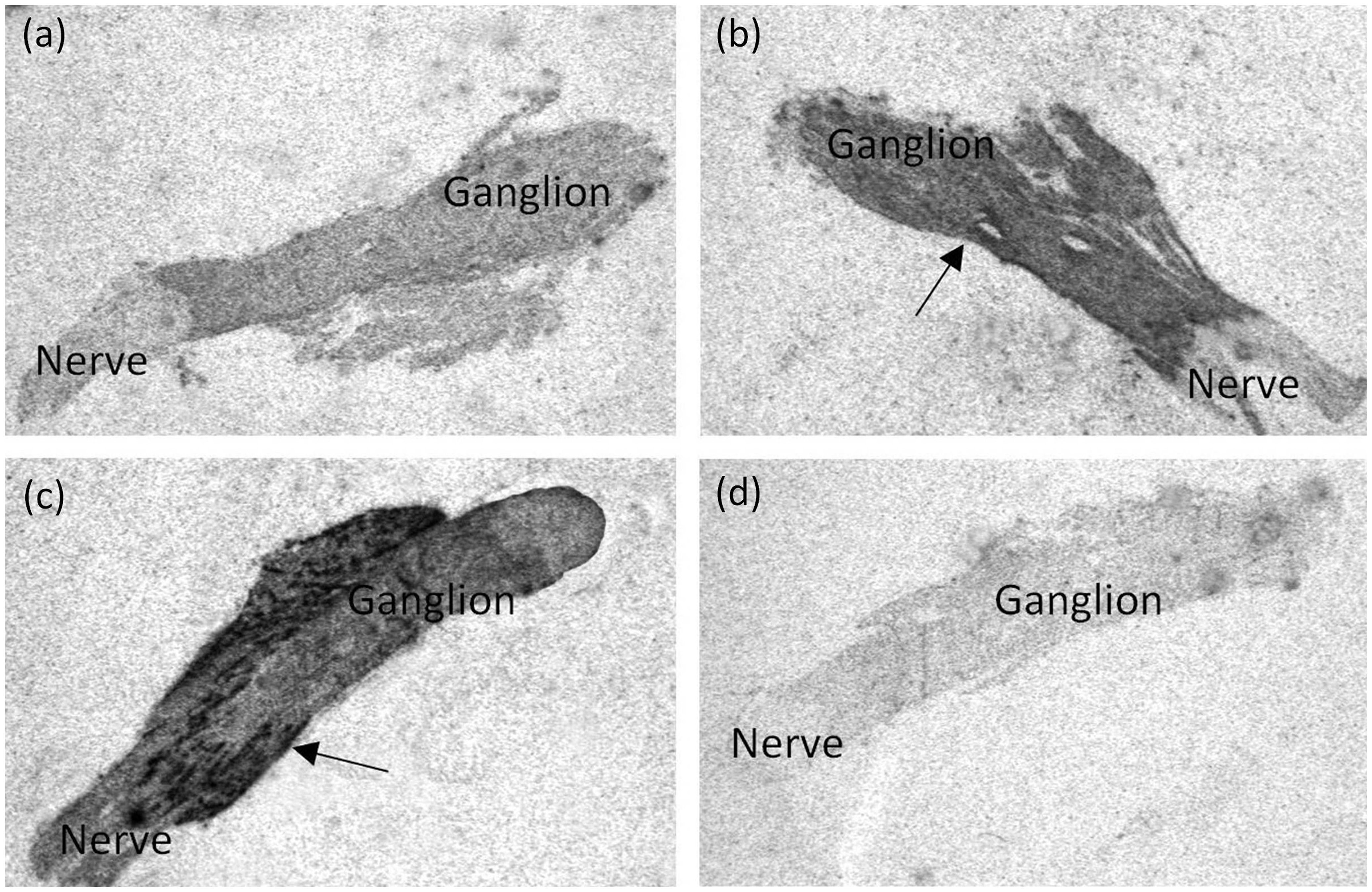

Changes in PDE activity by inhibitors of PDE3 and PDE5
Effects on cGMP hydrolysis
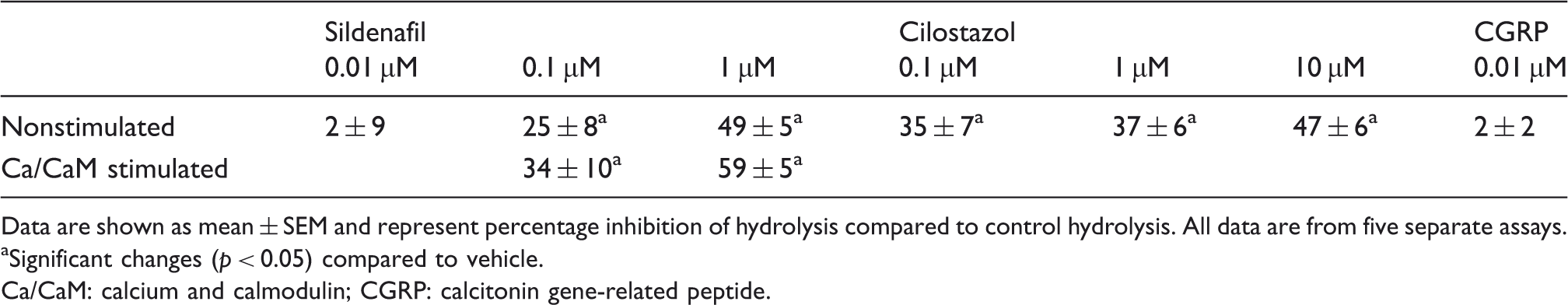
The activity of PDE3 and PDE5 was investigated in lysates of homogenized trigeminal ganglia. Compared to vehicle, addition of 0.1 µM of the PDE5 inhibitor sildenafil (Figure 5 and Table 3) caused a significant reduction of 34% (±6%) in cGMP hydrolysis. At higher concentrations of sildenafil (1 µM), the cGMP hydrolysis was significantly reduced 65% (±6%) compared to vehicle whereas lower concentrations of sildenafil (0.01 uM) caused no significant reduction in cGMP hydrolysis.
cGMP hydrolysis in response to migraine-relevant substances. Lysates from rat trigeminal ganglia were tested for PDE activity. PDE inhibitors or substances were added alone or in combinations as indicated. The results represent mean ± SEM of five experiments. Significant changes are indicated by bar and asterisk (*). Inhibition of total cGMP hydrolysis in percentage of control. Data are shown as mean ± SEM and represent percentage inhibition of hydrolysis compared to control hydrolysis. All data are from five separate assays. Significant changes (p < 0.05) compared to vehicle. Ca/CaM: calcium and calmodulin; CGRP: calcitonin gene-related peptide.

Although sildenafil is highly selective for PDE5 inhibition (IC50 = 3.5 nM), at high concentrations it can also inhibit PDE1 (IC50 = 281 nM (18)), which is stimulated by Ca/CaM and hydrolyzes cGMP. The activity of PDE1 and the effect of sildenafil on the PDE1-associated cGMP hydrolysis was investigated by concomitant addition of Ca/CaM to the lysate. At baseline, Ca/CaM enhanced cGMP hydrolysis, corresponding to an activation of PDE1; however, it was not a significant increase compared to vehicle without Ca/CaM. Addition of sildenafil (1 µM) significantly reduced cGMP hydrolysis by 68% (±10%) compared to vehicle. Thus, 1 µM sildenafil also inhibits PDE1.
A possible cross-talk between the cAMP and cGMP pathways was investigated by use of cilostazol. Cilostazol (0.1 µM, 1 µM, and 10 µM) did not significantly inhibit cGMP hydrolysis. In addition, CGRP had no effect on cGMP hydrolysis.
Effects on cAMP degradation
The effect of PDE inhibitors on cAMP degradation is shown in Figure 6 and Table 4. Cilostazol caused a significant decrease of 35% (±7%), 37% (±6%), and 47% (±6%) compared to vehicle in cAMP degradation at 0.1 µM, 1 µM, and 10 µM, respectively. The addition of CGRP did not elicit a significant effect on cAMP degradation. Interestingly, compared to vehicle, sildenafil (0.1 µM and 1 µM) caused a significant decrease in cAMP hydrolysis of 25% (±8%) and 49% (±5%). Adding 0.1 µM or 1 µM sildenafil and Ca/CaM also resulted in a significant reduction in hydrolysis of 34% (±10%) and 59% (±5%), respectively.
cAMP hydrolysis in response to migraine-relevant substances. Lysates from rat trigeminal ganglion were tested for PDE activity. PDE inhibitors or substances were added alone or in combinations as indicated. The results represent mean ± SEM of five experiments. Significant changes are indicated by bar and asterisk (*). Inhibition of total cAMP hydrolysis in percentage of control. Data are shown as mean ± SEM and represent percentage inhibition of hydrolysis compared to control hydrolysis. All data are from five separate assays. Significant changes (p < 0.05) compared to vehicle. Ca/CaM: calcium and calmodulin; CGRP: calcitonin gene-related peptide.
Discussion
This study clearly demonstrates that PDE3A, PDE3B, and PDE5A are each present in the trigeminal ganglion at the protein level and they are confined to neurons. In 20%–30% of the PDE3 and PDE5 immunoreactive neurons, the migraine-inducing peptide CGRP co-localized with the PDEs. Previous studies have reported approximately 30% CGRP-positive cells in the trigeminal ganglion (19,20), though other studies have reported up to 44% CGRP-positive cells. Such differences can probably be attributed to differences in antibodies and immunohistochemical procedures.
The in situ hybridization revealed a distribution of mRNA for all PDEs similar to the observed immunohistochemical stainings of the corresponding proteins, indicating that production and function of the PDEs are spatially identical.
Furthermore, we have demonstrated PDE3 and PDE5 activity in the trigeminal rat ganglion. This work also revealed a possible link between the cAMP and cGMP pathways in headache induction by PDE3 and PDE5 inhibitors because sildenafil influenced both cGMP and cAMP degradation.
In a human headache model, compounds inhibiting PDE3 and PDE5 elicit headache in healthy humans and migraine-like headache in patients suffering from migraine (5,6,21). This headache includes accompanying symptoms such as photo- and phonophobia. PDE inhibitors may mediate either a vascular or central effect in the pain pathway or a combination of both. Recent work has shown that extra-cranial arteries do not dilate during migraine headache (4), giving support to the neuronal hypothesis. The inhibitory effect of sildenafil on cAMP degradation indicates a possible dual cyclic nucleotide effect of sildenafil in the trigeminal ganglion at high concentrations in cerebral arteries as reported previously (22). Because both sildenafil and cilostazol can influence cAMP degradation, cAMP may be a key molecule in migraine pain, which is concordant with the headache-inducing effect of CGRP.
Trigeminal ganglion and role in headache pain
In pain conditions such as migraine, cluster headache, and trigeminal neuralgia, changes in trigeminal neuronal signaling are proposed to be involved. The primary sensory fibers of the trigeminal nerve are divided into three main branches: the ophthalmic, maxillary, and mandibular nerves, each of which provides somatosensory innervations of specific regions of the face and orofacial cavity (23). The trigeminal ganglion includes pseudounipolar neurons, Schwann cells, and satellite glial cells. Schwann cells associate with nerve fibers and produce myelin (24,25). The pseudounipolar neurons of the trigeminal ganglion are characterized by a single process that divides into a long peripheral and a shorter central branch. Two types of neurons are present in the trigeminal ganglion, large and small-to-midsize neurons. The populations overlap but have different characteristics. The large neurons have fast-conducting myelinated fibers whereas the small-to-midsize neurons have slower conducting C fibers, several of which contain neurotransmitters such as substance P and CGRP. These neurons are involved in thermo- and mechanoreception, and many of them are nociceptive (25–27). The cell bodies of the primary afferent neurons that convey sensory information from the periphery to the central nervous system are surrounded by several satellite glial cells, which number 10 times more than neurons in the trigeminal ganglion. Satellite glial cells surround individual sensory neurons and regulate the neuronal microenvironment, e.g. availability of nutrients and metabolites as well as neurotransmitters (24,28).
PDE3 and PDE5 are located in the trigeminal ganglion, placing them at the center of the pain signaling. Modulation of the cellular signaling in the ganglion could modulate the central sensing of pain.
PDEs and headache
The PDE families are characterized by their preferred substrate, cAMP or cGMP, as well as their mode of regulation. Each PDE may have a unique subcellular localization even within subtypes of the same PDE family (29). The PDE5A family consists of one gene and currently three identified splice variants (PDE5A1–PDE5A3). PDE5 is a cytosolic protein that hydrolyzes cGMP at low substrate concentrations and can be further activated by cGMP binding and phosphorylation by protein kinase G (29). The PDE3 family includes two genes, PDE3A and PDE3B. They are structurally similar, each containing a catalytic carboxy terminal and an amino terminal that is important for intracellular localization of the enzyme (30). Three splice variants for PDE3A have been identified (PDE3A1, PDE3A2, and PDE3A3); the A1 and A2 splice variants exist as membrane-bound proteins and A3 as a cytosolic protein (31). So far, no splice variants of the membrane-bound PDE3B have been identified (29,32). The PDE3 protein predominantly hydrolyzes cAMP, although it may also hydrolyze cGMP, with a lower Km for cGMP; the hydrolysis of cAMP is almost 10-fold higher than the Vmax for cGMP. In vitro cGMP can thus act as an inhibitor of cAMP degradation. In vivo, cGMP may inhibit PDE3, thereby earning its name as the “cGMP-inhibited phosphodiesterase” (30). The fact that PDE3 is inhibited by high levels of cGMP is evident in Figure 6, showing that the addition of sildenafil (high cGMP) results in a decrease in cAMP degradation. From the activity assays, it appears that cilostazol and sildenafil have a similar ability to augment levels of cAMP, which could suggest a common pathway both for sildenafil and cilostazol to induce headache through cAMP-related mechanisms.
In a previous study of human and guinea pig cerebral arteries, sildenafil likewise inhibited cAMP hydrolysis by 15%–26% (33), which is less than seen in the present study of approximately 50%. Whether such differences are related to species or tissue differences has not been established.
The hypothesis of a predominantly cAMP-mediated common pathway in headache induction is consistent with other headache-inducing molecules such as CGRP, which activates cAMP. The key relay point in headache may be cAMP signaling in the trigeminal ganglion, which should be confirmed by further investigations. Although cAMP has numerous targets downstream, the essential regulation of cAMP levels by PDE3 could be a principal modulator in the spatio-temporal setting of headache pathophysiology.
In conclusion, location and activity of PDE3 and PDE5 in the trigeminal ganglion place them at the center of neuronal signaling in the headache pain pathway. The new finding of an almost similar inhibition of cAMP hydrolysis in the rat trigeminal ganglion both by cilostazol and sildenafil suggests an indirect effect of sildenafil through inhibition of PDE3 by the induced increase in cGMP. This result suggests that cAMP might be a key relay molecule in the pain pathway; however, further testing in humans is needed. Activation of PDE3 or other related cAMP-selective PDEs could be a new target in migraine treatment alongside a selective PDE5 activator.
Clinical implications
Phosphodiesterases 3 and 5 are present and active in the neuronal part of the trigeminovascular system. Phosphodiesterases 3 and 5 are co-localized with calcitonin gene-related peptide in rat trigeminal ganglia. Both sildenafil and cilostazol can inhibit cAMP hydrolysis: cilostazol through direct inhibition and sildenafil by indirect inhibition. This study indicates that the key relay molecule in migraine could be cyclic adenosine monophosphate (cAMP). Activators of phosphodiesterases 3 and 5 could be future drug targets.
Footnotes
Funding
The study was supported by the Lundbeck Foundation (LUCENS), A.P. Møller Foundation for the Advancement of Medical Science, a Christian og Ottilia Brorsons travel grant, and the Cool Sorption Foundation of 1988.
Conflict of interest
None declared.