
Abstract
Objective
The objective of this article is to evaluate the efficacy and tolerability of two doses of oral sumatriptan vs placebo in the acute treatment of migraine in children and adolescents.
Background
Currently, there is no approved prescription medication in Japan for the treatment of migraine in children and adolescents.
Methods
This was a multicenter, outpatient, single-attack, double-blind, randomized, placebo-controlled, parallel-group study. Eligible patients were children and adolescents aged 10 to 17 years diagnosed with migraine with or without aura (ICHD-II criteria 1.1 or 1.2) from 17 centers. They were randomized to receive sumatriptan 25 mg, 50 mg or placebo (1:1:2). The primary efficacy endpoint was headache relief by two grades on a five-grade scale at two hours post-dose.
Results
A total of 178 patients from 17 centers in Japan were enrolled and randomized to an investigational product in double-blind fashion. Of these, 144 patients self-treated a single migraine attack, and all provided a post-dose efficacy assessment and completed the study. The percentage of patients in the full analysis set (FAS) population who report pain relief at two hours post-treatment for the primary endpoint was higher in the placebo group than in the pooled sumatriptan group (38.6% vs 31.1%, 95% CI: −23.02 to 8.04, p = 0.345). The percentage of patients in the FAS population who reported pain relief at four hours post-dose was higher in the pooled sumatriptan group (63.5%) than in the placebo group (51.4%) but failed to achieve statistical significance (p = 0.142). At four hours post-dose, percentages of patients who were pain free or had complete relief of photophobia or phonophobia were numerically higher in the sumatriptan pooled group compared to placebo. Both doses of oral sumatriptan were well tolerated. No adverse events (AEs) were serious or led to study withdrawal. The most common AEs were somnolence in 6% (two patients) in the sumatriptan 25 mg treatment group and chest discomfort in 7% (three patients) in the sumatriptan 50 mg treatment group.
Conclusions
There was no statistically significant improvement between the sumatriptan pooled group and the placebo group for pain relief at two hours. Oral sumatriptan was well tolerated.
Background
Migraine is a common and debilitating disorder in children and adolescents. Recently, Abu-Arafeh et al. (1) analyzed prevalence of headache including migraine in children and adolescents in a systematic review of 35 population-based studies of pediatric headache from 23 countries. The review showed that prevalence of migraine in the 3–19 years age group was 1.4%–13.8% (average: 6.0%) in males, and 2.1%–28.4% (average: 9.7%) in females, with the average prevalence of migraine of 7.7%. Other reports showed that the mean age of onset of migraine was 7 years for boys and 11 years for girls. Gender ratio changes with age. Migraine prevalence is greater in boys in the 3- to 7-year-old group. In ages 7–11 years, no significant difference in migraine prevalence between boys and girls was found. And at age 15, girls suffer from migraine more frequently (2,3). In earlier studies, diagnosis of migraine was made according to the classification and diagnostic criteria for headache disorders, cranial neuralgias and facial pain (International Headache Society (IHS): 1988) (4), and the International Classification of Headache Disorders, second edition (ICHD-II: 2004) (5) used in the trials conducted since 2006. In the Japanese study of adolescents aged 12–15 years (6), overall prevalence of migraine diagnosed according to ICHD-II was 4.8%, with 6.0% in boys and 9.7% in girls.
Establishing the correct diagnosis of migraine is crucial for adequate management. The treatment should be individually tailored and can include nonpharmacological and pharmacological approaches. While nonpharmacological treatment such as avoiding triggers and keeping a regular sleep schedule as well as a well-balanced diet can be helpful, pharmacological treatment of migraine attacks also may be needed.
Currently there is no approved prescription medication in Japan for the treatment of migraine in children and adolescents. The Japanese Practice Guidelines for Headaches, first edition (2007), states that ibuprofen and acetaminophen have been considered to be safe and effective for the acute treatment of pediatric migraine (7). Two studies supported the efficacy of ibuprofen and one study supported the efficacy of acetaminophen for the acute treatment of migraine in adolescents (8,9). However, ibuprofen is not licensed for treatment of migraine attacks in children and adolescents in Japan and is used off label.
Triptans are a class of drugs acting selectively as agonists on serotonin 5-HT1B/1D receptors. Sumatriptan, developed by GlaxoSmithKline for treatment of migraine, was the first clinically available triptan. Several studies of sumatriptan nasal spray have demonstrated efficacy and tolerability for acute treatment of migraine in children and adolescents (10–12), whereas the study of oral sumatriptan did not demonstrate clear efficacy (13). This difficulty might be attributed to a higher placebo response rate in children and adolescents as compared to adults (14–16). Several triptans have become available for migraine treatment in adults since 2000 in Japan; however, none of these has been licensed for use in children or adolescents.
The aim of the current study was to evaluate the efficacy, safety and tolerability of oral sumatriptan at doses of 25 mg and 50 mg for the acute treatment of a single migraine attack in children and adolescents. To date, there has been no study of triptan conducted in Japanese children and adolescents.
Methods
Ethical considerations
The study was conducted in accordance with Good Clinical Practice (GCP), Article 14-3 and 80-2 of the Pharmaceutical Affairs Law, all applicable patient privacy requirements, and the guiding principles of the Declaration of Helsinki (Edinburgh 2000, Washington 2002 and Tokyo 2004). The institutional review board/independent ethics committee reviewed and approved the study protocol and patient informed consent form. Written informed consent was obtained from each patient and his or her legal guardian prior to participation in the study.
Study design
This was a multicenter, outpatient, single-attack, double-blind, randomized, placebo-controlled, parallel-group study of two doses of oral sumatriptan vs placebo. This study was conducted from September 28, 2009 to December 3, 2010 in Japan.
Eligible patients were randomized to receive sumatriptan 25 mg, 50 mg or placebo. When randomization was completed, the patients entered a six-week treatment phase during which they could self-treat a single migraine attack according to the treatment arm. Study medication needed to be taken as soon as possible (within 0.5 hours) after the development of a migraine attack associated with grade 3 or more pain (moderate to severe pain) on the five-grade scale. The baseline for each migraine attack was the time the patient took the study medication. A 24-hour pain-free period prior to the onset of headache pain was required to establish that each migraine attack represented a new episode, rather than a recurrence of a previous headache.
Patients
Eligible patients were males and females 10–17 years old without childbearing potential or using adequate birth control having a minimum of a six-month history of migraine with or without aura as defined by ICHD-II criteria; had two to eight migraine attacks monthly for the two months prior to entry into the study; had migraine attacks associated with pain intensity of 3 or more on a five-grade scale that last a minimum of three hours; had shown no response to at least one nonsteroidal anti-inflammatory drug (NSAID) or acetaminophen; were able to distinguish migraine from other headaches; and were able to read, comprehend, and complete patient diaries.
Patients were ineligible for the study if they had weight <30 kg, ≥15 headache days per month in total (migraine, probable migraine, or tension-type headache), were suffering with retinal (ICHD-II 1.4), basilar (ICHD-II 1.2.6), hemiplegic (ICHD-II 1.2.4 or 1.2.5), or ophthalmoplegic migraine (ICHD-II 13.17); had secondary headaches, a history of cerebrovascular disease or myocardial infarction; signs or symptoms of cardiac ischemia; uncontrolled hypertension, peripheral vascular syndromes; epilepsy or structural brain lesions lowering convulsive threshold, history or manifestations of significant liver or kidney dysfunction were also excluded. In addition, patients were excluded if they used ergot medications in the previous three months for migraine prophylaxis or had changed dose of any other medication for migraine prophylaxis within two months prior to randomization; had taken or planned to take a monoamine oxidase inhibitor anytime within the two weeks prior to entry into the study, or had psychotropic drug, alcohol or substance abuse within the last year.
Blinding and randomization
The investigator informed potential patients and the legal guardians of this study and obtained consent from both. Following this, screening tests and examinations were performed, and if the patient satisfied all eligibility criteria, the investigator (or subinvestigator) completed the registration form and sent it to the randomization center by facsimile. The randomization center ensured that the registration form contained all necessary information and that the patients satisfied all eligibility criteria of the protocol and assigned a randomization number to the patient. For allocation of the participants, a computer-generated list of random numbers was used. The investigator (or subinvestigator) then dispensed the investigational product to the patient according to the computer-generated randomization number. The patients were randomized to sumatriptan 25 mg or 50 mg or placebo in 1:1:2 ratios using random block sizes of four. The sites were trained on how to maintain blind conditions.
Study assessments
To measure efficacy, patients were required to complete diaries at baseline and at regular intervals through four hours post-treatment. Pain intensity was evaluated by using the five-grade categorical rating scale on which no pain, mild, mild to moderate, moderate to severe, and severe pain were scored as 1, 2, 3, 4 and 5, respectively. Date and time of onset, presence of migraine-associated symptoms, such as photophobia, phonophobia, nausea and vomiting were recorded in the diaries.
The primary efficacy endpoint was the percentage of patients who reported pain relief (defined as at least a two-point reduction on the five-grade scale) at two hours post-treatment without previous use of rescue medication.
Secondary efficacy endpoints were the percentage of patients who reported pain relief at time points at 0.5, one and four hours; the percentage of patients who were pain free at 0.5, one, two and four hours post-treatment; the percentage of patients who were free of each of the following symptoms: photophobia, phonophobia, nausea, or vomiting at 0.5, one, two, and four hours post-dose as well as the percentage of patients who used rescue medication from the time of dosing to four hours post-dose. Pain-free was defined as a pain intensity score of “1” (no pain) in patients who had not used rescue medication prior to the assessment. If rescue medication was taken, for subsequent efficacy assessments pain intensity was assumed to be severe and the symptoms were set to be present for the purpose of the data analysis. Rescue medication could include either a single oral dose of an NSAID or acetaminophen, not exceeding the maximum recommended single dose or an antiemetic. Safety evaluation included AEs, laboratory assessments (hematology, blood chemistry and urinalysis), vital signs and electrocardiogram (ECG). Severe adverse events (SAEs) were collected from the time of informed consent up to and including any follow-up contact. AEs were collected from the start of the investigational product until the follow-up contact.
Statistical analysis
Based on the randomization ratio of 1:1:2 (sumatriptan 25 mg, 50 mg and placebo), a total of 140 patients were required to test the hypothesis at the two-sided significance level of 0.05 with 80% power; 166 patients were needed for randomization, assuming a drop-out rate of 15%. Calculations are based on Fisher’s exact test. The percentages of patients who would report pain relief at two hours for sumatriptan and placebo groups were assumed as 64% and 39%, respectively, based on the data from a published nasal sumatriptan pediatric trial (9). A pre-defined analysis plan was followed with no interim analysis planned or performed.
The safety population (SP) consisted of all patients who took at least one dose of the investigational product. The full analysis set (FAS) consisted of all patients in the SP who provided any post-treatment efficacy assessment. The per protocol set (PPS) consisted of all patients in the FAS who did not deviate from any major protocol requirements. The last observation carried forward (LOCF) dataset was defined as the dataset imputed by the LOCF method. Only the post-treatment value was used for the imputation. The observed case (OC) dataset was defined as the dataset without any missing data imputation. The OC dataset was used for a part of the efficacy analysis. These analyses were secondary and aimed to confirm the stability of LOCF results.
SAS® software was also used to perform all calculations. The primary comparison of interest was the percentage of patients who reported pain relief at two hours in the sumatriptan pooled group (pooled across doses) vs placebo groups. The comparison was performed with a significance level of 0.05 (two sided) and based on FAS and LOCF datasets. Other comparisons were two sided and carried out at the significance level of 0.05. However, no adjustment for multiplicity was made since they were secondary comparisons.
The primary efficacy analysis was based on FAS and LOCF datasets. The number and percentage of patients who reported pain relief at two hours were summarized for sumatriptan pooled and placebo groups. The difference in percentages between both groups was calculated along with 95% confidence intervals, and the statistical comparison was made by χ2 test. The null hypothesis for this study was that there is no difference between sumatriptan pooled and placebo groups in the percentage of patients who report pain relief at two hours. The alternative hypothesis was that there is a difference between sumatriptan pooled and placebo groups in the percentage of patients who report pain relief at two hours.
Secondary analyses were conducted for pain relief, pain free and rescue medication use. The number and the percentage at each assessment time point (0.5, one, two and four hours) or period of “0–4 hours” for rescue medication use were summarized for each treatment (sumatriptan 25 mg, 50 mg, sumatriptan pooled, and placebo). The differences in percentages between each sumatriptan group vs placebo were calculated along with 95% confidence intervals, and the statistical comparisons were made by χ2 test. Similar analyses as pain relief and pain free were performed for associated symptoms (nausea, vomiting, photophobia and phonophobia). Only patients who had each associated symptom at the treatment were included in the denominator.
Any AEs occurring within the time period that extended from the date of investigational product use up to (and including) the follow-up contact were summarized. The summaries of AEs both by body weight and by age were produced. Summary statistics (n, mean, standard deviation, median, minimum and maximum) were provided at each scheduled time point. Also, the number and percentage of patients with data outside normal ranges were tabulated at each scheduled time point. The number and percentages of each urinalysis test result were displayed at each scheduled time point. Summary statistics (n, mean, standard deviation, median, minimum and maximum) for vital signs and physical examinations were provided at each scheduled time point. The number and percentages for ECG findings were displayed.
Results
Study population
A total of 178 patients from 17 centers in Japan were enrolled and randomized to an investigational product in double-blind fashion (Figure 1). Of these, 144 patients self-treated a single migraine attack; all provided a post-dose efficacy assessment and completed the study. All 144 patients provided a post-treatment efficacy assessment and were included in the FAS population. Given that all the 144 patients were evaluated for safety and tolerability, the SP and FAS datasets were identical. A total of 136 patients in the FAS population completed the study without any major protocol violations and were included in the PPS population. Eight subjects in the FAS population had major protocol deviations and were therefore excluded from the PPS population (four patients were taking concomitant medication prohibited by the protocol. The other four patients had noncompliance in taking the investigational product).
Flowchart of patient disposition.
Demographic and baseline characteristics (SP population).

SP: safety population; Min: minimum; Max: maximum; SD: standard deviation.
Two patients had both migraine types.
Efficacy findings
Pain relief
The percentage of patients in the FAS population who reported pain relief at two hours post-dose (the primary endpoint) was higher in the placebo group than in the sumatriptan pooled group (38.6% and 31.1%, respectively; Figure 2(a)) but failed to achieve statistical significance (p = 0.345). The percentage of patients in the FAS population who reported pain relief at four hours was numerically higher in the sumatriptan pooled group than the placebo group (sumatriptan pooled: 63.5%, placebo: 51.4%; Figure 2(a)) but failed to achieve statistical significance (p = 0.142). Similar results were observed in the PPS population (placebo: 39.4%, sumatriptan pooled: 31.4, p = 0.331) and were also seen when analysis by body weight or age was performed (Figure 2(b)).
(a) The percentage of patients with pain relief at 0.5, one, two and four hours post-treatment (full analysis set (FAS) population). (b) The percentage of patients with pain relief at two hours post-treatment by age (FAS population).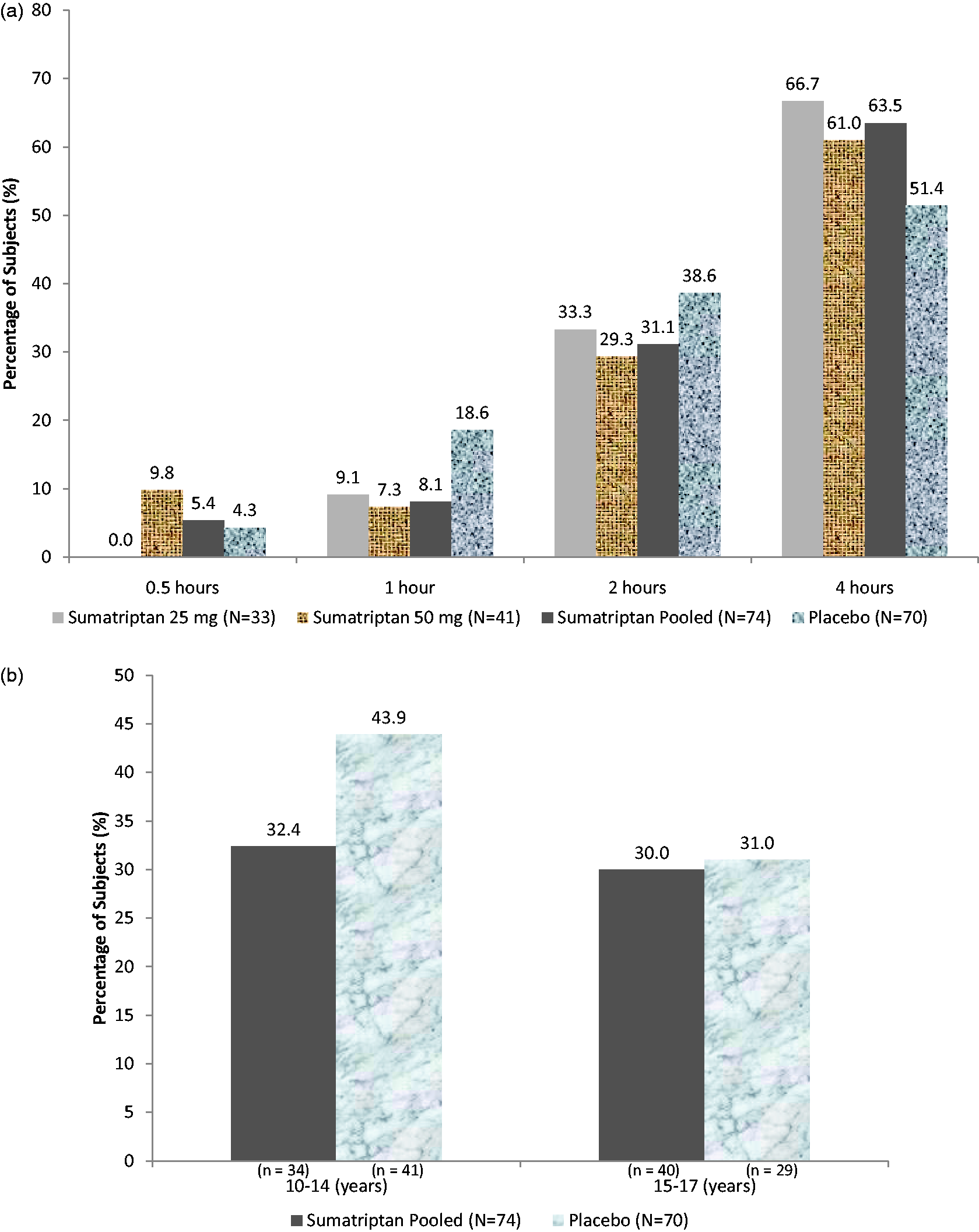
Pain free
The percentage of patients in the FAS population who were pain free at 0.5, one, and two hours post-dose was numerically higher in the placebo group than in the sumatriptan pooled group. The percentage of patients who were pain free at four hours was numerically higher in the sumatriptan pooled group than in the placebo group (sumatriptan pooled: 50.0%, placebo: 47.1%; Figure 3). Statistical significance was not reached for the sumatriptan pooled group compared to the placebo group (p = 0.732).
The percentage of patients who were pain free at 0.5, one, two, and four hours post-treatment (full analysis set (FAS) population).
Associated symptoms
The percentage of patients who were photophobia free at two hours was numerically higher in the sumatriptan pooled group than the placebo group (sumatriptan pooled: 53.6%, placebo: 52.8%). No statistically significant difference was observed between the placebo group and the pooled sumatriptan group for this symptom (p = 0.950). The percentage of patients who were phonophobia free at two hours was numerically higher in the placebo group than the sumatriptan pooled group (placebo: 63.6%, sumatriptan pooled: 53.3%). Similarly, a numerically greater percentage of patients was nausea free in the placebo group vs the pooled sumatriptan group (81.0% and 61.1%, respectively). At four hours post-dose, the proportions of patients who had complete relief of photophobia or phonophobia were numerically higher in the sumatriptan pooled group compared to placebo. No statistically significant difference was observed between the placebo group and the pooled sumatriptan group at this time point.
Use of rescue medication
The use of rescue medications was comparable across all treatment groups (12.1%, 14.6%, 13.5%, and 12.9% in the sumatriptan 25 mg, 50 mg, sumatriptan pooled groups and placebo, respectively).
Safety findings
Adverse event summary (SP).
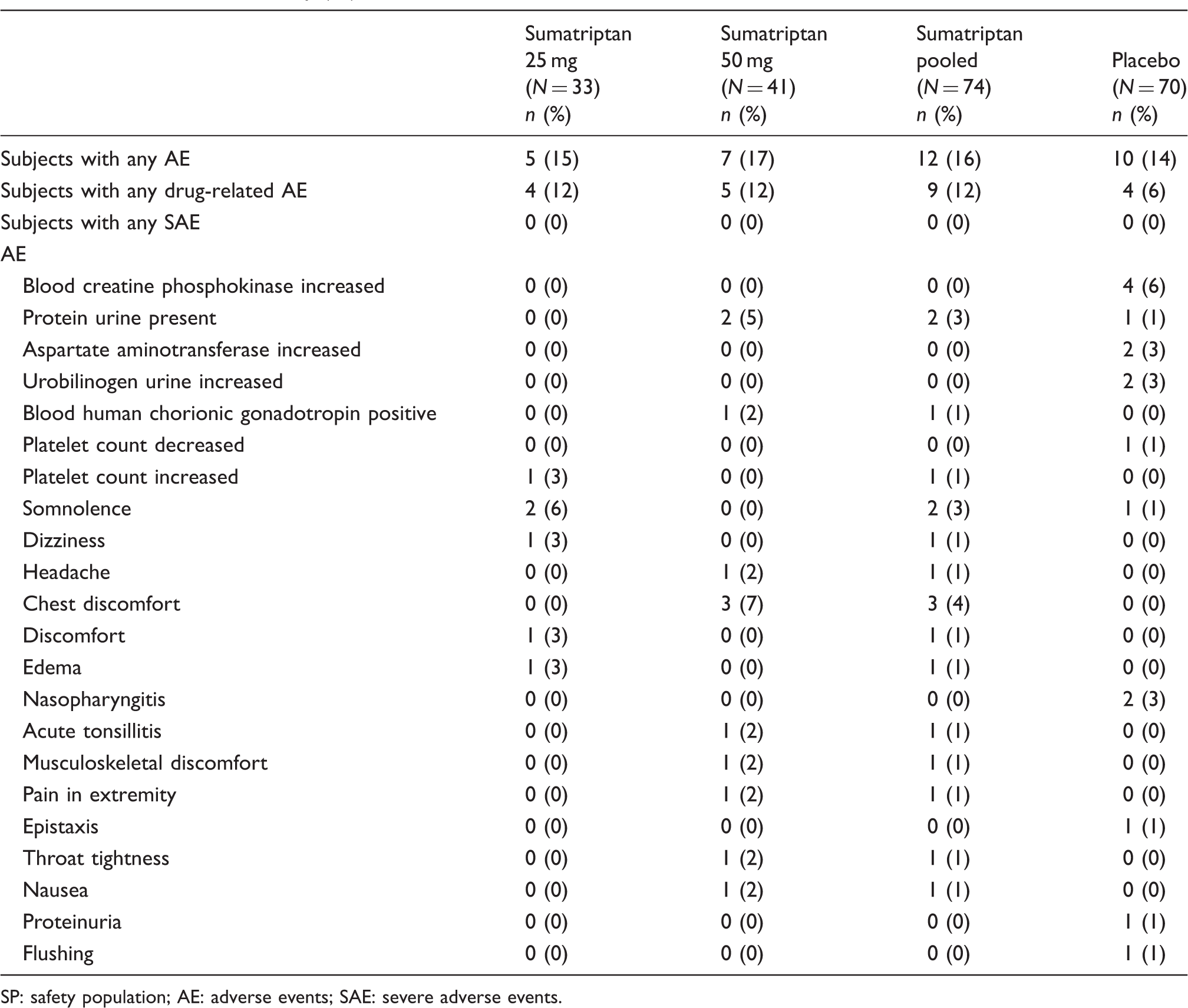
SP: safety population; AE: adverse events; SAE: severe adverse events.
Discussion
Children with headaches are usually referred to general pediatrics or neurological clinics; however, this category of patients seems to be underrepresented among the referrals to pediatric neurologists in Japan as compared with other neurologic disorders such as epilepsy syndromes, myopathies and developmental disorders. Migraine is the most frequent primary headache in the outpatient setting because of its severity and associated manifestations.
Accurate diagnosis according to ICHD-II is the first step for migraine treatment. Nonpharmacological treatment such as avoiding triggers, and regular sleep and exercise are recommended for pediatric migraine. If pharmacological treatment is needed, analgesics like ibuprofen and acetaminophen are usually administered. However, even if taken in recommended doses and early in the course of the attack, these medications can be ineffective in severe migraine attacks in some patients.
There is a considerable body of research focusing on pharmacological management of pediatric migraine; however, data from controlled trials are still limited and therefore pharmacological treatment of migraine in children and adolescents is mainly off label (3,7). For acute treatment of migraine, the most rigorously studied agents are ibuprofen, acetaminophen and selected triptans (rizatriptan and almotriptan tablets, sumatriptan and zolmitriptan nasal sprays) (3). In our experience, the efficacy of sumatriptan nasal spray is favorable but the bad taste is not tolerated by some children with migraine, therefore tablet formulation may be preferred by some young migraineurs. However, to date, no study of a triptan has been conducted in children and adolescents in Japan.
This was the first randomized, double-blind, placebo-controlled trial of oral sumatriptan to evaluate its efficacy and tolerability in the treatment of migraine in Japanese children and adolescents. In the current study, patients in the sumatriptan pooled group experienced lower pain relief than in the placebo group at two hours.
The high placebo response in acute treatment of migraine in children and adolescents has been reported in a number of trials (14–16) whereas low placebo response rate was observed in a recent study (17). A parallel-group study design and the use of a four-point pain scale were suggested as the determinants of the high placebo effect (15,16). The placebo response rate in pediatric migraine was shown to be higher than that in adult migraine in the systematic review (15). Factors associated with a lower placebo response rate in children and adolescent trials of acute treatment of migraine were single-center (vs multicenter) trials, and small sample size. Placebo response rate did not depend on age and gender (16).
Our study was a parallel-group multicenter trial from 17 headache centers, and pain relief as a primary endpoint was defined as at least a two-grade reduction on a five-grade scale, which is the same for the sumatriptan nasal spray study that indicated significant efficacy (11). The inclusion of 17 centers might be one of the reasons why this study failed to demonstrate efficacy. However, it was uncertain why pain relief at two hours in the sumatriptan pooled group was lower than that in the placebo group despite the same pain-relief definition as the study for sumatriptan nasal spray. The time to onset of action for sumatriptan nasal spray was 15 minutes. For the sumatriptan tablet, the time was 30 minutes (18). The difference of the time to onset might be one of the reasons why sumatriptan nasal spray is effective but sumatriptan in tablet form is not. A recent oral rizatriptan study has indicated that the rizatriptan was significantly more effective than the placebo and well tolerated for migraine attacks in patients older than 6 years (19). One of the reasons why the study indicated significant efficacy could be the appropriate selection of an outcome measure to evaluate pain intensity. Headache diaries used in the study were completed by the children and also by parents proving assessment of the child’s behavior (18). In contrast, headache diaries in our study were filled in only by patients. We used two doses of sumatriptan (25 mg and 50 mg), but did not use 100 mg, because in Japan, 50 mg is the standard dose for adults. Therefore, we used only both 50 mg and 25 mg. However, the use of low-dose sumatriptan (25 mg and 50 mg) might be one of the reasons why this study failed to demonstrate efficacy.
We used a single dose without the option of a repeat dose after two hours because the duration of pediatric migraine is shorter than the duration of adults’ migraine. Therefore, we think that treatment by only a single dose is sufficient for Japanese children. Actually 66.7% of subjects treated by placebo in the study achieved pain relief.
We treated one single attack, not three attacks in this trial, because it made study participation easier for Japanese patients; treating three attacks is great burden for the Japanese patients. However, some patients may not respond to sumatriptan in some, but not all, migraine attacks.
The recent sumatriptan and naproxen sodium combination tablets study (17) employed a number of strategies to reduce placebo response rates and improve treatment difference detection, including a definitive primary endpoint (percentage of pain-free patients) that did not require assessing degree of pain; treatment at moderate/severe pain for greater post-treatment pain differentiation; a placebo run-in phase to identify and exclude placebo responders; a selection of subjects with migraines attacks lasting >3 hours to avoid the impact of natural resolution; and site selection emphasizing the pediatric experience for improved communication with adolescents.
Our study did not reach statistical significance on the primary endpoint with the percentage of patients reporting pain relief at two hours port-dose being numerically higher in the placebo group vs the pooled sumatriptan group.
Although analysis of secondary efficacy endpoints in this study generally also did not show a treatment effect, patients in the sumatriptan pooled group experienced numerically higher pain relief; photophobia, phonophobia and were pain free (but had no nausea) compared to the placebo group at four hours post-dose.
The overall incidence of AEs in each treatment group was lower than the rates observed for equivalent doses in previous studies in adults (20). These results suggested that oral sumatriptan for acute treatment of migraine was well tolerated in children and adolescents. No clear treatment effect was observed in the AEs by age and weight. The results should be interpreted with caution because of the small number of subjects in the subcategories. The results of this study showed that oral sumatriptan was less effective in children and adolescents than in adults. However, a proportion of young migraineurs can benefit from oral sumatriptan at four hours post-dose, but characteristics of this subset of the patient population are not known. Based on the literature, such characteristics of the trial as parallel-group design, the use of outcome measure assessing degrees of pain as well as completion of diaries only by subjects could have contributed to the high placebo response rate.
Further studies to evaluate the efficacy and safety of sumatriptan in children and adolescents are be needed, and these studies should incorporate strategies to minimize placebo response.
Conclusions
There was no statistically significant improvement between the sumatriptan pooled group and the placebo group for the primary endpoint, pain relief at two hours. At four hours post-dose, the proportions of patients who experienced pain relief, were pain free, or had complete relief of photophobia or phonophobia were numerically higher in the sumatriptan pooled group compared to placebo. The most frequently reported AEs were somnolence and chest discomfort in the sumatriptan 25 mg group and the sumatriptan 50 mg group, respectively. No SAEs were reported. Oral sumatriptan was well tolerated for the treatment of migraine in children and adolescents.
Clinical implications
There was no statistically significant improvement between the sumatriptan pooled group and the placebo group for the primary endpoint, pain relief at two hours. At four hours post-dose, the proportions of patients who experienced pain relief, were pain free or had complete relief of photophobia or phonophobia were numerically higher in the sumatriptan pooled group compared to placebo. Oral sumatriptan was well tolerated for the treatment of migraine in children and adolescents.
Footnotes
Acknowledgment
This study was sponsored by GlaxoSmithKline (Tokyo, Japan), the manufacturer of oral sumatriptan. The investigators received compensation for conducting the study. K. Sato and H. Nishioka are employees of GlaxoSmithKline.
Funding
This work was supported by GlaxoSmithKline.
Conflicts of interest
K. Sato and H. Nishioka are employees of GlaxoSmithKline, the license holder of oral sumatriptan. M. Fujita has been a clinical consultant/advisor to GlaxoSmithKline for this study. F. Sakai has been an investigator in this study.