
Abstract
Background
BMS-927711 is a potent, selective, competitive human calcitonin gene-related peptide (CGRP) receptor antagonist that has shown in vivo efficacy without vasoconstrictor effect. The objective of the current study was to determine an effective and tolerable dose range of BMS-927711 for the acute treatment of migraine.
Methods
In this randomized, double-blind, placebo controlled, dose-ranging study, 885 patients were randomized using an adaptive design to one of the following dose groups: BMS-927711 (10, 25, 75, 150, 300, or 600 mg); sumatriptan 100 mg (active comparator); and placebo. Patients were treated for a single migraine attack. The primary endpoint was pain freedom at two hours post-dose.
Results
Of patients who took the study drug, 799 had one post-randomization efficacy evaluation. Significantly more patients in the BMS-927711 75 mg (31.4%, p = 0.002), 150 mg (32.9%, p < 0.001), and 300 mg (29.7%, p = 0.002) groups and the sumatriptan group (35%, p < 0.001) had pain freedom at two hours post-dose versus placebo (15.3%). For the secondary endpoint of sustained pain freedom from two to 24 hours post-dose, BMS-927711 doses (25–600 mg) were also statistically significant compared with placebo. No deaths or treatment-related serious adverse events (AEs) were reported, and no patients discontinued because of AEs.
Conclusions
BMS-927711 is superior to placebo at several different doses (75 mg, 150 mg, and 300 mg) and has an excellent tolerability profile.
Introduction
Only one class of acute migraine-specific medication—serotonin 5-HT1B/1D receptor agonists: triptans—has been developed and approved for the treatment of migraine over the past two decades. The introduction of triptans represented a shift toward drugs more selectively targeting the suspected pathophysiology of migraine. While triptans have been a boon to many patients, issues such as an incomplete effect or headache recurrence remain important clinical limitations (1,2). In fact, only about one-third of patients from clinical trials are pain free at two hours after taking triptans (1). In addition, they are contraindicated in patients with cardiovascular diseases, cerebrovascular disease, or significant risk factors for either because of potential systemic and cerebrovascular vascoconstriction from the 5-HT1B-mediated effects (3). Thus, there remains a significant unmet medical need for migraine-specific medication without cardiovascular liability.
Calcitonin gene-related peptide (CGRP) plays an important role in migraine pathophysiology, and may provide a novel mechanism for acute migraine treatment (4). Animal models have shown that stimulating trigeminal sensory fibers evokes CGRP-mediated vasodilation of dural vessels (5). Migraine attacks are associated with increased CGRP serum concentrations (6), and CGRP infusions can trigger attacks in individuals with migraine (7). CGRP serum concentrations can be reversed after triptan administration, an effect that coincides with migraine symptom relief (8). Initial clinical studies show that CGRP receptor antagonists are effective in aborting migraine attacks (9–11). presumably by one of several putative mechanisms, including blocking CGRP-induced neurogenic vasodilation (12), or inhibiting either trigeminocervical nociceptive transmission or thalamic trigeminal nociceptive activation (13,14) or both.
BMS-927711 is a potent, selective, competitive human CGRP receptor antagonist (15). This report presents the results of a double-blind, randomized, multicenter, outpatient evaluation of the safety, efficacy, and dose response of six different oral doses of BMS-927711 as compared to placebo in the treatment of moderate or severe migraine headache. The purpose of this study was to evaluate the efficacy of BMS-927711 compared with placebo in the acute treatment of migraine as measured by pain freedom at two hours post-dose, while identifying an optimal dose to support the Phase 3 clinical trials.
Methods
This was a single-dose, double-blind, randomized, multicenter, outpatient evaluation of BMS-927711 as compared to placebo in the treatment of moderate to severe migraine headache. In addition, oral sumatriptan at 100 mg was used in this trial as the active-control for assay sensitivity. The study, which lasted from October 2011 to May 2012, was divided into three phases: a screening/baseline phase (three to 28 days), an acute treatment phase (up to 45 days during which time subjects treated one migraine headache of moderate to severe intensity), followed by an end-of-treatment visit within seven days of administration of study medication. Patients were randomized to receive placebo, sumatriptan (100 mg), or one of six doses of BMS-927711: 10 mg, 25 mg, 75 mg, 150 mg, 300 mg, or 600 mg (Figure 1).
Study design.
Patients returned to the study site within seven days of study treatment for review of the electronic diary, assessment of medication compliance, and monitoring of tolerability (adverse events (AEs)) and safety (including vital signs, laboratory tests, and electrocardiography (ECGs)). The relatively rapid outcome measure of an acute migraine trial: two hours post-dose following a single episode of migraine attack, presents an opportunity to utilize an adaptive dose-finding design to assess a wide range of doses (16). By facilitating dynamic treatment allocations based on information accumulated, an adaptive design allowed more efficient sample size distribution between doses and a full characterization of the dose response. Furthermore, by implementing early stopping rules in an adaptive design for either success or futility, potential limitations could be achieved with respect to patient exposure to ineffective treatment(s), providing further efficiency in total sample size.
This study was conducted in accordance with the ethical principles that have their origin in the current Declaration of Helsinki, and was consistent with International Conference on Harmonization Good Clinical Practice and applicable regulatory requirements. The study protocol and any amendments were reviewed and approved by the Institutional Review Board/Independent Ethics Committee for each site prior to initiation of the study. This trial is registered with ClinicalTrials.gov (ID: NCT01430442).
Inclusion criteria
The study included male and female subjects, 18 to 65 years of age, who had at least a one-year history of migraine with or without aura. In addition, the migraine should have started prior to 50 years of age, with an average duration of about four to 72 hours if untreated. Patients also were required to have between two and seven attacks of moderate to severe intensity in each of the three months prior to the screening visit. Patients were to have less than 15 days with headache per month in each of the three months prior to the screening visit and during the screening period, and had to be able to distinguish migraine attacks from other headache types, such as tension-type. Preventive migraine medication was permitted while on study therapy provided the dose was a stable dose for at least three months prior to study entry. Serotonin norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors, and monoamine oxidase inhibitors required a 14-day wash-out period prior to study entry.
Exclusion criteria
In view of the sumatriptan arm, patients with a history of basilar-type migraine or hemiplegic migraine were excluded from the study, as were patients who did not receive migraine relief from triptans. Patients were excluded if they had a history or evidence of stroke/transient ischemic attacks, ischemic heart disease, coronary artery vasospasm, other significant underlying cardiovascular diseases, uncontrolled hypertension (high blood pressure), uncontrolled diabetes, or human immunodeficiency virus (HIV) disease. Patients with a current diagnosis of major depression, other pain syndromes, psychiatric conditions (e.g. schizophrenia), dementia, or significant neurological disorders, other than migraine, that in the investigators’ opinion might interfere with study assessments, were excluded. Patients with a history of, treatment for or evidence of alcohol or drug abuse within the past 12 months or patients who have met Diagnostic and Statistical Manual of Mental Disorders, fourth edition text revision (DSM-IV-TR) criteria for any significant substance use disorder within the past 12 months from the date of the screening visit were excluded.
Concomitant drugs metabolized by CYP3A with narrow therapeutic margin with theoretical potential for drug interaction were not permitted in this study, as potent CYP3A inhibitors and inducers could cause changes in the pharmacokinetics of BMS-927711. Medications that may alter the pH of the stomach, such as H2 receptor antagonists, proton pump inhibitors, and antacids, were prohibited. The use of barbiturates, opioids, triptans, ergotamines, and muscle relaxants were prohibited two days prior to randomization and during the course of this study.
Women of child-bearing potential who were unwilling or unable to use an acceptable contraceptive method or abstinence to avoid pregnancy for the entire study period and for up to eight weeks after the study were not permitted to enroll, as were women who were pregnant or were breastfeeding. Women with a positive pregnancy test on enrollment or prior to study drug administration were excluded.
Sample size, randomization, and treatment
In the absence of an early stop, the study was to randomize roughly 1100 patients, of whom it was expected that approximately 825 patients would be treated. Patients were randomized to receive one of six doses of BMS-927711 (10 mg, 25 mg, 75 mg, 150 mg, 300 mg, or 600 mg), sumatriptan (100 mg), or placebo as a single dose, once the subject’s migraine headache qualified as moderate to severe in intensity. No dose adjustments were permitted. Patients who did not experience relief of their migraine headache at the end of two hours after dosing with study medication were permitted to use the following rescue medication(s): aspirin, ibuprofen, acetaminophen, a nonsteroidal anti-inflammatory drug (NSAID), anti-emetics, or baclofen. However, patients could take their prescribed standard-of-care medications for treatment of migraine if it was 48 hours after administration of the one dose of study medication, and before coming in for the end-of-treatment visit, provided all of the assessments were completed on the electronic handheld subject diary.
Randomization schedules were generated and kept by the study sponsor. Each subject who qualified for treatment was assigned a unique randomization number via an interactive voice response system. Study medication (four capsules) was dispensed via a boxed kit that contained three 95 cc bottles with one capsule in two of the bottles and two capsules in one of the bottles. Subjects were given the study medication kit at randomization (baseline visit) and were instructed to take all four capsules from the kit at the time of moderate to severe migraine headache onset.
Allocation of subjects was accomplished in two phases, a fixed allocation and an adaptive algorithm, similar to those utilized in other migraine trials (9,11,16,17). The trial initially ran like a fixed-dose, parallel-groups design. In this design, 336 subjects were initially to be randomized in fixed ratios to each of the treatment arms. A total of 84 patients were allocated to placebo, and 36 were allocated to each of the remaining arms. This “burn-in” phase provided substantial data about the efficacy of the treatment groups prior to implementing the adaptive algorithm. In the adaptive phase of this trial, two out of every eight patients were randomized to placebo, one of every eight were randomized to sumatriptan, and the remaining five patients were adaptively allocated to the BMS-927711 treatment arms. After the first phase, allocation to the BMS-927711 dose groups was adaptively determined once per week by a Bayesian analysis of the observed response rates. The Bayesian algorithm flexibly allocated subjects to treatment groups based on information that was accumulated as the trial progressed. As the dose-response relationship began to be elucidated by the data, the algorithm dynamically allocated more subjects to the doses of BMS-927711 that were likely to be of clinical interest and fewer subjects to the less efficacious doses. Thus, the number of subjects allocated to each of the BMS-927711 doses was not known until the end of the study. An interim analysis was performed for administrative purposes after 600 subjects were treated.
Outcomes
The primary objective was to evaluate the efficacy of BMS-927711 compared with placebo in the acute treatment of migraine as measured by pain freedom: headache pain intensity level reported as “no pain,” at two hours post-dose using a four-point rating scale (no pain, mild pain, moderate pain, severe pain). Patients who experienced relief of headache pain to a mild intensity or pain-free intensity level at two hours post-dose were considered to be responders. The patients who did not experience such relief at the end of two hours were permitted to use an approved rescue medication. Use of rescue medication within 48 hours was also recorded. Whatever the case, the patient was required to continue to complete his or her electronic diary for up to 48 hours after consuming the study medication.
Secondary efficacy variables included total migraine freedom: a composite endpoint consisting of freedom from headache pain coupled with no symptoms of photophobia, phonophobia, and nausea, at two hours post-dose, and sustained pain freedom from two to 24 hours. Exploratory measures included pain relief: no pain or mild pain, at two hours post-dose, sustained pain relief from two to 24 hours post-dose, freedom from photophobia, phonophobia or nausea at two hours post-dose, and sustained pain freedom from two to 48 hours post-dose.
Safety variables included AEs, serious adverse events (SAEs), clinical laboratory evaluations, vital sign measurements, physical examinations, and ECGs.
Statistical analyses
The primary efficacy endpoint was the proportion of pain-free subjects at two hours post-dose. The primary efficacy analysis was based on a Bayesian, hierarchical, logistic model of the dose-response relationship of the primary endpoint and used the Last Observation Carried Forward (LOCF) data set of all subjects in the efficacy population. The primary efficacy endpoint was evaluated both for statistical significance and for clinical superiority using a comparison of the dose estimated to be the ED90 versus placebo. The definitions of these two criteria were:
Statistical significance: Posterior probability that the ED90 dose was better than placebo was at least 95%–i.e. Pr(ED90 > Placebo) ≥0.95. Clinical superiority: Posterior probability was at least 90% that the response rate for the ED90 dose was at least 15 percentage points greater than the response rate for placebo–i.e. Pr(ED90–Placebo > 15%) >0.90.
A sensitivity analysis, which also compared the ED90 to placebo, was carried out on the primary efficacy endpoint, where the percentage of pain-free subjects was evaluated by the Cochran Mantel Haenszel (CMH) General Association test controlling for treatment group (BMS-927711 ED90 dose versus placebo) and baseline pain severity. The null hypothesis that there was no difference between the two treatment arms with respect to the primary endpoint was tested with a two-sided test at a 0.05 significance level. A dose-response analysis that compared each treatment arm to placebo, using uncorrected CMH tests, was also conducted.
The secondary efficacy endpoint of total migraine freedom was a composite score, set to “yes” if pain freedom and no symptoms of photophobia, phonophobia, and nausea were observed, at two hours post-dose. This endpoint was evaluated using the same analytical methods (primary and sensitivity) described above for the primary endpoint.
The secondary endpoints of sustained pain freedom from two to 24 hours and from two to 48 hours, and sustained pain relief from two to 24 hours, were analyzed using a CMH model and the LOCF data set. For the associated symptoms of nausea, phonophobia, and photophobia, ad hoc analyses were conducted that used uncorrected CMH tests to compare each treatment arm to placebo. The secondary safety analyses for AEs and SAEs summarized the number of subjects who had one or more events by treatment group. The events were also listed by treatment arm.
Results
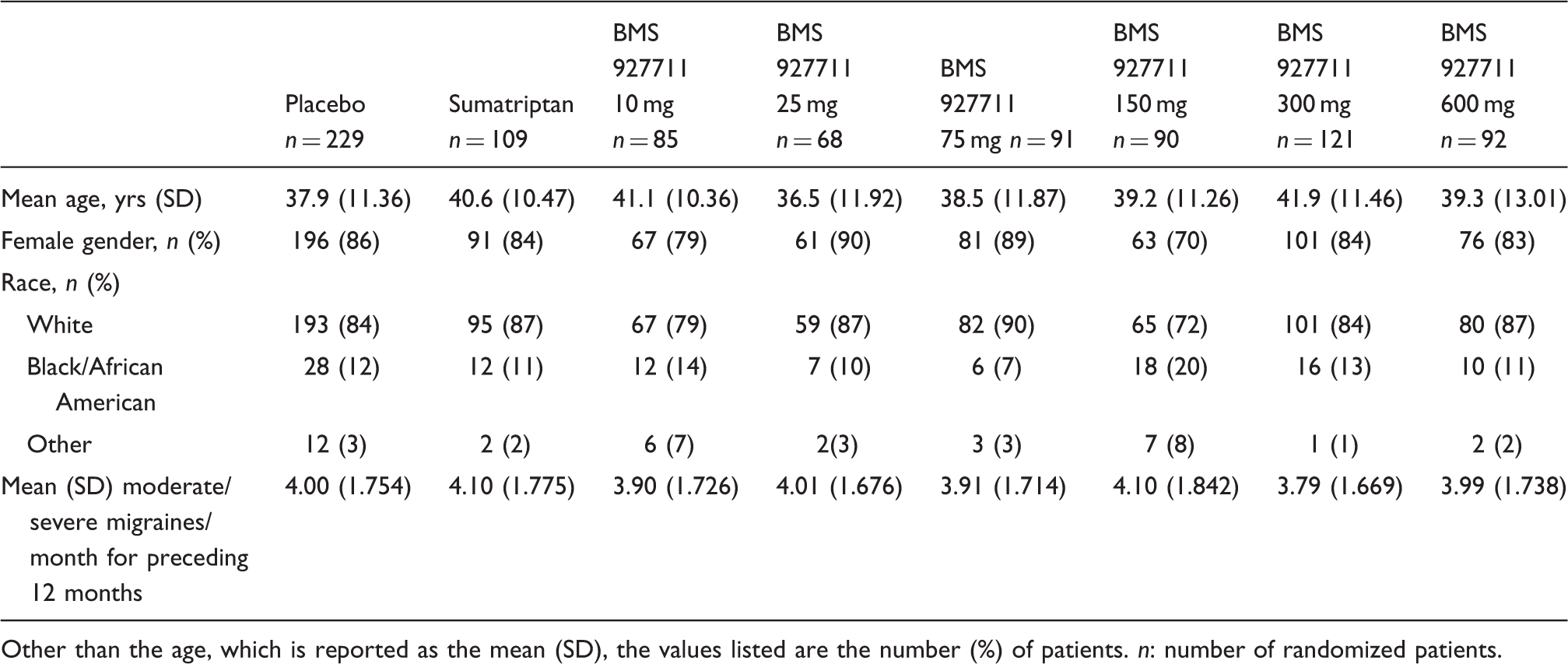
A total of 1026 patients were enrolled in the study, and 86.3% (885) of these were randomized to treatment. Of the 885 randomized subjects, 812 (79.1%) completed the study (Figure 2). The baseline demographic characteristics of the randomized patients are listed in Table 1.
Patient disposition. Patient baseline demographic characteristics by dosing strategy (all randomized patients). Other than the age, which is reported as the mean (SD), the values listed are the number (%) of patients. n: number of randomized patients.
“No longer met criteria” means that the patient did not treat a moderate to severe migraine attack. “Completed study” means that the patient treated the migraine attack and performed all of the follow-up assessments. n: sample size.
For purposes of analysis, the safety population, defined as randomized patients who took at least one capsule of study medication, included 811 patients. The efficacy population, defined as all subjects in the safety population who had at least one post-randomization efficacy evaluation and a corresponding baseline pain evaluation for the treated headache, included 799 patients.
Primary efficacy endpoint
The percentage of patients who were pain free at two hours post-dose is depicted in Figure 3. The Bayesian model showed that among the BMS-927711 doses, the 150 mg dose was the most effective, with 32.9% of subjects achieving pain freedom at two hours post-dose. The 150 mg dose was significantly better than placebo for pain freedom at two hours post-dose (p < 0.001). The difference in response rates between the 150 mg dose and placebo was not significantly larger than 15% (p = 0.28) at that time point.
Primary pain endpoint: Percentage of patients who were pain-free at two hours post-dose.
In comparison with the placebo group, a dose-response curve, with maximum efficacy at 150 mg, was demonstrated. Black bars: patients in the BMS-927711 dose arms; gray bars: patients in the sumatriptan arm. ap < 0.05.
Sumatriptan and the 75 mg, 150 mg, and 300 mg BMS-927711 dose groups each were significantly superior to placebo (p < 0.001 for sumatriptan; p ≤ 0.002 for the BMS dose groups) for the percentage of patients with pain freedom at two hours post-dose. Among the BMS-927711 dose groups, the percentage of subjects who were pain free at two hours post-dose was 32.9% (28/85) in the 150 mg; 31.4% (27/86) in the 75 mg; and 29.7% (33/111) in the 300 mg dose, compared to 15.3% of patients in the placebo group. No additional benefit was seen with the 600 mg dose (24.4%, 20/82).
Secondary efficacy endpoints: Total migraine freedom and sustained pain freedom (two to 24 hours)
The percentage of patients who experienced total migraine freedom: no pain, nausea, photophobia, or phonophobia at two hours post-dose, is depicted in Figure 4. The Bayesian model showed that among the BMS-927711 doses, the 75 mg dose was the most effective, with 28.2% of subjects achieving total migraine freedom at two hours post-dose. The 75 mg dose was significantly better than placebo (p < 0.001). The difference in response rates between the 75 mg dose and placebo was not significantly larger than 15% (p = 0.64) at this time point.
Secondary pain endpoint: Percentage of patients who achieved total migraine freedom at two hours post-dose.
In comparison with the placebo group, a dose-response curve, with maximum efficacy at 75 mg, was demonstrated. Black bars: patients in the BMS-927711 dose arms; gray bars: patients in the sumatriptan arm. ap < 0.05.
Sumatriptan and the 75 mg, 150 mg, and 300 mg groups each were significantly more effective than placebo (p ≤ 0.007) in achieving total migraine freedom at two hours post-dose. Among the BMS-927711 dose groups, the percentage of subjects who had total migraine freedom at two hours post-dose was 27.9% (24/86) in the 75 mg; 25.9% (22/85) in the 150 mg; and 23.4% (26/111) in the 300 mg doses, compared to 11.8% of patients in the placebo group.
Summary of efficacy for key secondary and exploratory endpoints.
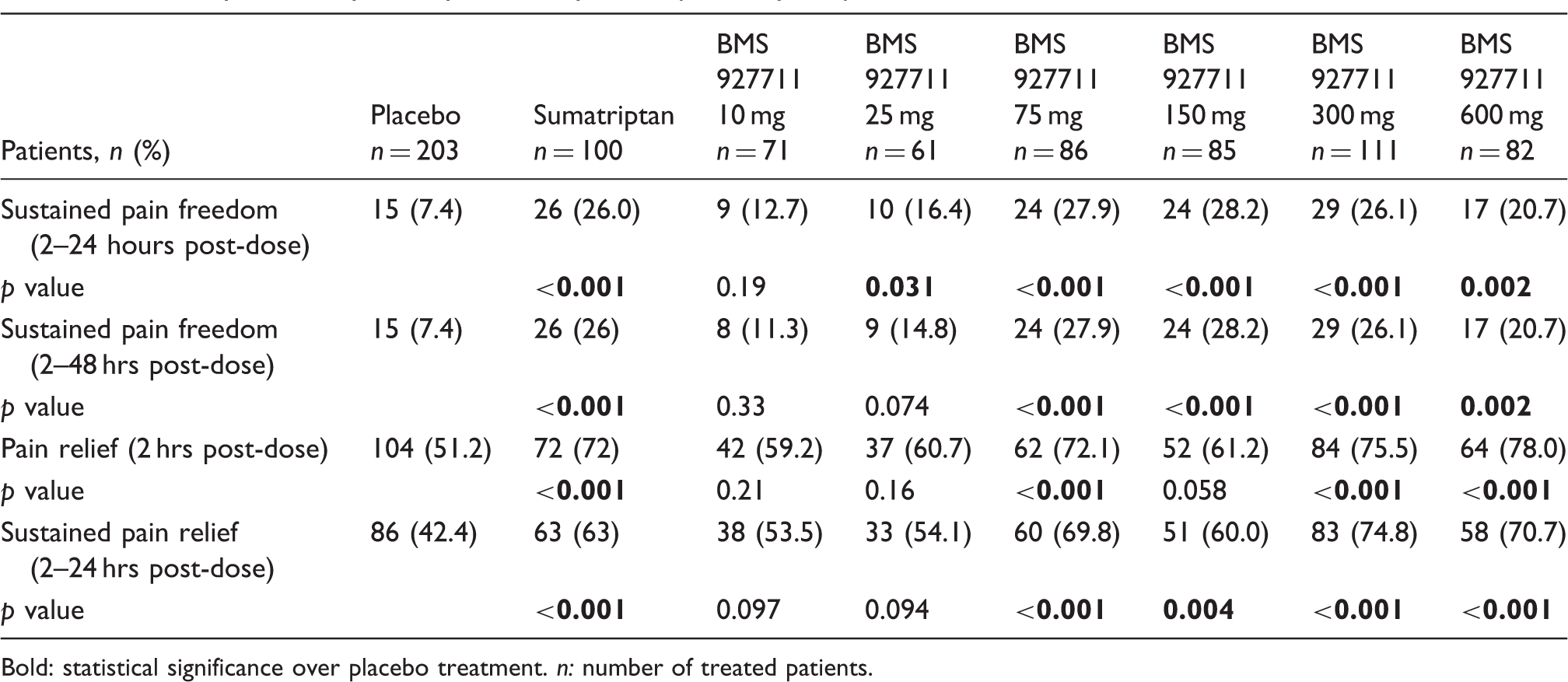
Bold: statistical significance over placebo treatment. n: number of treated patients.
Exploratory efficacy endpoints: Pain relief, sustained pain relief and sustained pain freedom (two to 48 hours)
The percentage of patients with pain relief: no pain or mild pain, at 2 hours post-dose, sustained pain relief (no recurrence of moderate or severe migraine and no use of rescue medication) from two to 24 hours, and sustained pain freedom from two to 48 hours, is listed in Table 2. The percentage of patients meeting each endpoint was significantly greater than placebo for the sumatriptan group and for multiple doses of BMS-927711. The percentage of subjects with pain relief continued to increase through 24 hours post-dose in all active treatment groups. By 48 hours post-dose, pain relief occurred in more than 88.5% of all patients.
Additional exploratory efficacy endpoints: photophobia, phonophobia, and nausea
Summary of efficacy of treatment on three associated migraine symptoms at two hours post-dose.
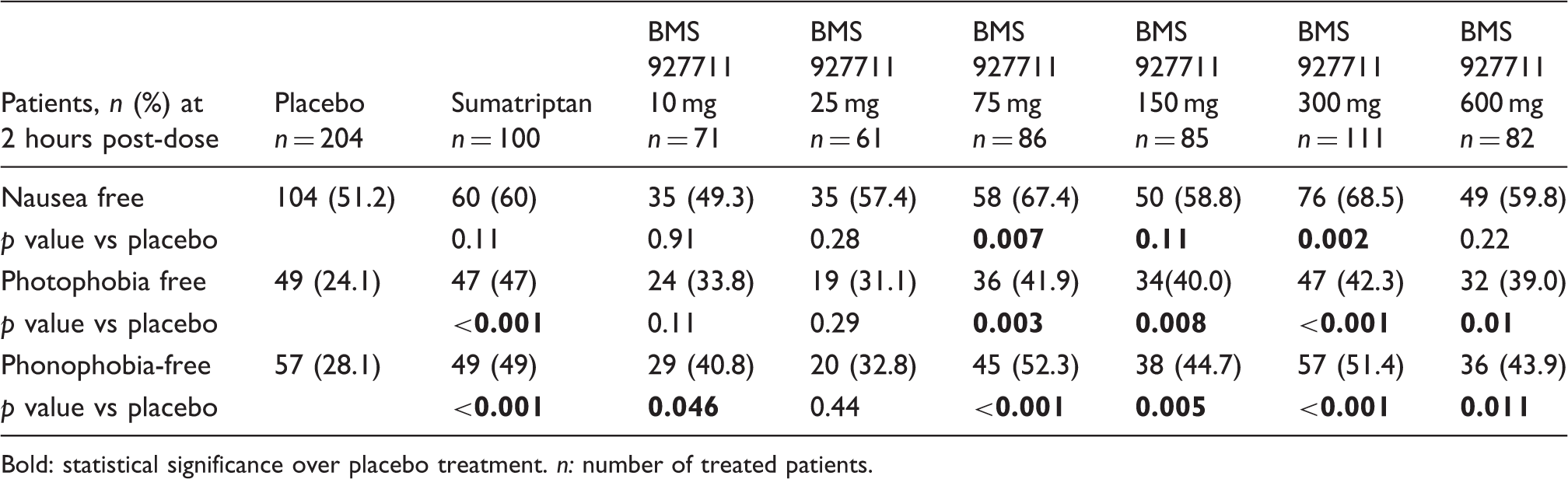
Bold: statistical significance over placebo treatment. n: number of treated patients.
By two hours post dose, the percentage of subjects without nausea was significantly greater than that of placebo in the BMS-927711 300 mg and 75 mg groups (Table 3). The difference between the placebo and sumatriptan group was not statistically significant at this time point. Improvement in nausea continued in all groups through 24 hours post-dose, with the sumatriptan, 75 mg, and 300 mg dose groups having the greatest percentages of subjects with total relief of nausea. By 48 hours post-dose, most subjects reported no nausea (data not shown).
Rescue medication
Overall, 35.5% (288/811) of patients took rescue medication during the study. Rescue medication was used most often in the placebo group (50.7%, 106/209), followed by the 600 mg (41.7%, 35/84); 10 mg (38.9%, 28/72); sumatriptan (31.0%, 31/100); 25 mg (29.0%, 18/62); 150 mg (25.6%, 22/86); 75 mg (24.4%, 21/86); and the 300 mg (24.1%, 27/112) arms. Overall, the most common rescue medications used during the study were ibuprofen (13.1%, 106/811) and acetaminophen/aspirin/caffeine (10.2%, 83/811).
AEs
Number (%) of patients reporting a commonly occurring a adverse event within 48 hours post-dose.

Adverse events occurring in ≥2% of patients in any treatment group; ordered by frequency in the BMS 927711 600 mg group. n: number (%) of patients who took at least one tablet of the study medication.
Of note, two patients had increased hepatic enzymes reported as an adverse event. One patient in the BMS-92771 75 mg dose group had a report of a mild increase in a hepatic enzyme on Day 7 that resolved after 64 days. The second patient was in the placebo group. No patients had an alanine aminotransferase (ALT) elevation that was ≥3 × upper limit of normal (ULN). One patient in the placebo group had a total bilirubin level that was ≥2 × ULN.
Performance of the adaptive design
The adaptive randomization scheme allocated subjects to the doses that were most efficacious in terms of pain freedom at two hours post-dose. The 75 mg, 150 mg, 300 mg, and 600 mg dose groups were allocated the highest number of subjects (n = 86, 85, 111, and 82, respectively). However, the subject allocation was not proportional to the final observed outcome. The 150 mg dose was, by the end of the study, numerically, slightly more efficacious than the 300 mg dose (28.2% versus 26.1%, respectively.) Yet, the 300 mg dose group received more subjects. This was because the 300 mg dose performed well early on in the study. This early advantage faded later in the study, but not in time to change the relative over-allocation of subjects to the 300 mg dose group. The 600 mg dose had relatively low efficacy (20.7% pain freedom at two hours), but received almost as many subjects (n = 82) as the 150 mg dose group. This was because of the properties of the Bayesian algorithm that smoothed the dose-response curve. Although the observed response rate dropped off at the highest dose, the smoothed dose-response curve fit by the algorithm was influenced by the behavior of the lower doses. Hence, it estimated the response rate of the 600 mg dose group to be higher than the observed rate.
Discussion
Here we report a demonstration of efficacy in acute migraine using the two-hour pain-free endpoint for the novel CGRP receptor antagonist BMS-927711. Pain-free rates ranged from 29.7% to 32.9% with a 15.3% placebo rate. The pain-free rate at two hours for sumatriptan 100 mg was 35%, which, taken with the somewhat high placebo response rate, illustrates the study had a broadly standard population. BMS-927711 was well tolerated, and as expected18 there were no cardiovascular issues. There remains an unmet medical need for nonvasoconstrictor antimigraine treatments, and BMS-927711 is a novel, orally available CGRP receptor antagonist without activity at the 5-HT1B receptor (19), allowing it to be a potentially new form of acute treatment for migraine.
In the evaluation of a new drug treatment, both efficacy and risk are important factors to assess. This trial demonstrated that multiple doses of BMS-927711 (75 mg, 150 mg, and 300 mg) were significantly more effective than placebo, demonstrating better pain freedom from migraine pain at two hours post-dose. The 600 mg dose was not superior to placebo on the primary endpoint, and this result may be because of the inherent variability present in the patients randomized to this dose group.
BMS-927711 was shown to be more efficacious than placebo in several secondary endpoints, although this study was not specifically powered to detect differences in these outcomes. One secondary endpoint evaluated sustained pain freedom from two to 24 hours post-dose. It demonstrated that 25 mg, 75 mg, 150 mg, 300 mg, and 600 mg doses of BMS-927711 were statistically significant over placebo. With respect to three different exploratory endpoints evaluating associated symptoms of nausea, photophobia, and phonophobia, several different doses of BMS-927711 were superior to placebo. For example, with respect to phonophobia freedom, 10 mg, 75 mg, 150 mg, 300 mg, and 600 mg were all superior to placebo. For photophobia treatment, the doses were slightly different; only 75 mg, 150 mg, 300 mg, and 600 mg were superior to placebo. Only the 75 mg and 300 mg doses of BMS-927711 were superior to placebo with respect to nausea freedom. Finally, two exploratory endpoints, pain relief at two hours post-dose and sustained pain relief from two to 24 hours post-dose, both indicate that there are several different efficacious doses of BMS-927711. Therefore, these data, taken together, support the idea that the 75 mg, 150 mg, and 300 mg doses of BMS-927711 are effective in the acute treatment of migraine pain.
Sumatriptan (100 mg) was used as an active control for assay sensitivity. This trial was not designed with the statistical power to allow comparison of the sumatriptan arm against the BMS-927711 treatment arms; therefore, efficacy between these groups cannot be assessed. However, this trial did demonstrate that sumatriptan was more effective than placebo with respect to the primary endpoint of pain freedom.
This study further supports the potential utility of CGRP receptor antagonists, which have generally exhibited efficacy in published clinical trials, for the acute treatment of migraine (9–11,17,20). The responses associated with BMS-927711 were comparable with other CGRP receptor antagonists tested in clinical trials. Sixty-six percent of patients treated with 2.5 mg olcegepant, an intravenously administered CGRP antagonist, exhibited pain relief two hours post-infusion in a Phase II migraine study, with 44% of patients being headache free at that time and dose (11). Similarly, 55% of patients with migraine treated with telcagepant, an orally administered CGRP receptor antagonist, experienced pain relief two hours post-dose in a Phase III migraine study, with 27% being pain free, and approximately 23% achieving total migraine freedom at two hours post-dose (10). Similarly, 21.9% of patients treated with 400 mg BI 44370 TA were migraine free at two hours post-dose in a Phase II study (20). Approximately 24%–36% of patients treated with the highest doses of MK-3207, a more potent CGRP receptor antagonist than telcagepant, were pain free two hours post-dose in a Phase II study (17).
There are limitations to the conclusions that can be drawn from this study. The single-dose design provides limited information regarding safety and tolerability, and it is not clear if the safety and efficacy profile would be similar with repeated use. There were no overt signs of liver toxicity in the current study, but the single-dose design and exclusion of drugs metabolized by CYP3A4 may not have provided sufficient exposure to uncover hepatic toxicity that would be evident with repeated use over time. For example, the development of another CGRP receptor antagonist compound was presumably halted because of elevated liver function tests with repeated use (21). A more complete assessment of hepatic effects with repeated dosing would need to be included in a Phase III study design. While the mechanism of action of CGRP receptor antagonists suggests that they may have fewer vasoconstrictor liabilities compared to triptans, the population in this study excluded patients with a history of specified cardiovascular conditions, making it impossible to determine if BMS-927711 would be safe or tolerable in such a population. In studies with another CGRP receptor antagonist, telcagepant, even when sought in patients with cardiovascular disease, no untoward effects have been identified (22,23).
In conclusion, this large Phase IIb trial identified multiple doses (75 mg, 150 mg, and 300 mg) of BMS-927711 that were superior to placebo with respect to a variety of efficacy endpoints. The effective BMS-927711 doses appeared at least comparable to sumatriptan though the study was not powered to detect differences between active treatments. Single doses of BMS-927711 were well tolerated, with the most common AEs including nausea, dizziness, and vomiting. Although additional information is needed with respect to longer-term use, the study successfully identified doses that could be carried forward for continued development.
Clinical implications
BMS-927711, a potent, selective, competitive human calcitonin gene-related peptide (CGRP) receptor antagonist, was assessed for the acute treatment of migraine in a randomized, double-blind, parallel-group, placebo-controlled, dose-ranging study that utilized an adaptive design. Patients were assigned to BMS-927711 (10, 25, 75, 150, 300, or 600 mg); sumatriptan 100 mg (active comparator); or placebo. Significantly more patients in the BMS-927711 75 mg (31.4%, p = 0.0018), 150 mg (32.9%, p = 0.005), and 300 mg (29.7%, p = 0.0024) groups had pain freedom at two hours post-dose vs placebo (15.3%). No deaths or treatment-related serious adverse events were reported, and no patients discontinued because of adverse events. BMS-927711 is superior to placebo with respect to a variety of efficacy endpoints at several different doses (75 mg, 150 mg, and 300 mg) and has an excellent tolerability profile.
Footnotes
Funding
This study was supported by Bristol-Myers Squibb.
Conflicts of interest
Drs Marcus, Stock, Manos, and Fischer are current or former employees of Bristol-Myers Squibb. Dr Goadsby is on advisory boards for Allergan, Colucid, MAP pharmaceuticals, Merck, eNeura, Neuraxon, Autonomic Technologies Inc, Boston Scientific, Electrocore, Eli Lilly, Medtronic, Linde gases, Arteaus, AlderBio, and Bristol-Myers Squibb. Dr Goadsby has consulted for Pfizer, Nevrocorp, Lundbeck, Zogenix, Impax and Dr Reddy, and has been compensated for expert legal testimony. Dr Goadsby has grant support from Allergan, Amgen, MAP, and Merck. Dr Goadsby has received honoraria for editorial work from Journal Watch Neurology and for developing educational materials and teaching for the American Headache Society. Dr Dodick has consulted for Allergan, Amgen, Arteaus, Ethicon, Rinat, Colucid, ENeura, NuPathe, Huntsworth, Medlogix, Alder, WL Gore, Y&R, Boston Scientific, Pfizer, Merck, Bristol-Myers Squibb, and MAP. Dr Dodick has received an honorarium from Sage Journals for editorial services.
Acknowledgments
Dr Stock supervised the statistical analyses and takes responsibility for the accuracy of the data. The authors would like to acknowledge Beth Morris and Tamara Bratt for their invaluable assistance as protocol managers, Timothy McCormack and Jennifer Stillman for their assistance as site managers, and Brian Atkinson, PhD, for writing and editorial assistance.
The authors would like to thank the investigators and study sites participating in the CN17003 study: Eugene Andruczyk, Philadelphia, PA, USA; Sheena K. Aurora, Seattle, WA, USA; Gary D. Berman, Minneapolis, MN, USA; Roger Cady, Springfield, MO, USA; Shane Glade Christensen, Salt Lake City, UT, USA; Leonard H.S. Chuck, Walnut Creek, CA, USA; Stephen E. Daniels, Austin, TX, USA; Matthew G. Davis, Rochester, NY; USA; Eugene Du Boff, Denver, CO, USA; Victor A. Elinoff, Endwell, NY, USA; John E. Ervin, Kansas City, MO, USA; Alex Feoktistov, Chicago, IL, USA; David L. Fried, Warwick, RI, USA; Ira J. Goodman, Orlando, FL, USA; Brian Grosberg, Bronx, NY, USA: Gregory Haase, Virginia Beach, VA, USA; Rashmi Halker, Scottsdale, AZ, USA; Wayne Harper, Raleigh, NC, USA; Dan C. Henry, Salt Lake City, UT, USA: John D. Hudson, Austin, TX, USA; Bruce Kohrman, South Miami, FL, USA; Richard A. Krause, Chattanooga, TN, USA; David Bruce Kudrow, Santa Monica, CA, USA; Robert S. Lipetz, Spring Valley, CA, USA; David Davis Meyer, Winston-Salem, NC, USA; Margarita Nunez, St. Petersburg, FL, USA; Antoinette A. Pragalos, Cincinnati, OH, USA; Anthony D. Puopolo, Milford, MA, USA; Harvey Resnick, Lake Jackson, TX, USA; Jay Rubin, Ocala, FL, USA; Joel R. Saper, Ann Arbor, MI, USA; David J. Seiden, Pembroke Pines, FL, USA; Gerald Shockey, Mesa, AZ, USA; Stephen D. Silberstein, Philadelphia, PA, USA; Timothy Smith, St Louis, MO, USA; Egilius Spierings, Watertown, MA, USA; Stuart R. Stark, Alexandria, VA, USA; Jon Christopher Stringer, Manlius, NY, USA; Cynthia Becher Strout, Mt Pleasant, SC, USA; Stewart J. Tepper, Cleveland, OH, USA; and Randal Lee Von Seggern, Greensboro, NC, USA.