
Abstract
Background
Glutamate is implicated in migraine pathophysiology; amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor antagonists represent a potential therapeutic approach because of their anti-excitatory actions.
Methods
This randomized, double-blind, proof-of-concept study assessed the efficacy of the AMPA receptor antagonist, BGG492 (250 mg), vs placebo and sumatriptan (100 mg), in 75 subjects with acute migraine attacks. Efficacy was measured using the Patient Migraine Diary. Pharmacokinetic and safety data were collected.
Results
Improvement from severe/moderate to mild/no headache pain (primary response) was reported in 58%, 58%, and 54% of BGG492-treated subjects at 2, 3, and 4 hours post-dose (p = 0.2, 0.5, and 0.5 vs placebo), respectively, compared with 68%, 84%, and 92% sumatriptan-treated subjects, and 40%, 48%, and 44% in the placebo group. Percentages of subjects with ≥ 2-point improvement in pain score from baseline at 2 hours were 29%, 40%, and 16% for BGG492, sumatriptan, and placebo, respectively. Pain-free response at 2 hours was reported for 25%, 24%, and 16% of BGG492, sumatriptan, and placebo subjects, respectively. Adverse events were reported by 80%, 56%, and 60% of BGG492, sumatriptan, and placebo subjects, respectively.
Conclusions
Proof-of-concept criterion was not met (≥ 25% BGG492 subjects with a primary response vs placebo at two timepoints). BGG492 was comparable to sumatriptan in terms of pain-free response.
Introduction
Migraine is a burdensome primary headache disorder (1). The introduction of the triptans, e.g. sumatriptan, has improved the treatment of acute migraine attacks (2). However, sumatriptan does not always abort migraine attacks; 100 mg of sumatriptan led to a reduction from moderate to severe to no pain at 2 hours in only one-third of patients in clinical trials (2), and did not provide headache relief in almost one-third of patients in general practice (3). In addition, triptans show mild vasoconstriction and so are contraindicated in migraine patients with vascular diseases, e.g. myocardial infarction, transient ischemic attack
Migraine pain is transmitted via the trigeminal nerve to the primary relay center for orofacial pain, the trigeminal nucleus caudalis (TNC) (5). Neuronal impulses in the trigeminal nerve are mediated by several neurotransmitters, including the major excitatory transmitter, glutamate (5). Receptors for glutamate are expressed in the trigeminal nerve and TNC, and are implicated in the main pathophysiological features of migraine (5,6). These include cortical spreading depression (CSD), trigeminovascular activation, and central sensitization (6). Pain sensitization occurs following periods of prolonged neuronal activity. This phenomenon results in excessive activation of sensory neurons and is thought to be mediated, in part, by amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, which are ionotropic glutamate receptors (6). Regulation of glutamate levels may also be linked to a genetic risk factor for migraine; a recent genome-wide association study in migraine sufferers identified a sequence variant on chromosome 8q22.1, which is flanked between two genes involved in glutamate homeostasis (7). One promising therapeutic approach for the management of migraine attacks is to block AMPA receptors in the pain pathway; this would decrease excitatory transmission and subsequent sensitization, thus reducing pain and allodynia. Blockade of AMPA receptors by the non-N-methyl-D-aspartate (NMDA) excitatory amino acid receptor blocker, GYKI 52466, produced a dose-dependent inhibition of trigeminovascular-evoked responses in the trigeminocervical complex of cats (8). Furthermore, the AMPA/kainate receptor antagonist, LY293558, has demonstrated therapeutic benefits in acute migraine (9).
BGG492 is an orally active, competitive antagonist of the AMPA receptor with high affinity for rat and human AMPA receptors (half maximal inhibitory concentration values (IC50) of 0.19 µM and 0.2 µM, respectively) and does not show AMPA receptor subtype selectivity in functional calcium influx assays. On native neocortical neurons, BGG492 shows a 145-fold selectivity for the AMPA receptor over the NMDA receptor. Compared with LY293558 (IC50 AMPA 1.35 μM, and kainate 4.8 μM), BGG492 is more selective for AMPA receptor (IC50 AMPA 0.186 μM) and has no affinity for the kainate receptors (data on file by Novartis). Considering the contribution of glutamate to migraine pathophysiology and the observed effects of AMPA antagonists in preclinical and clinical trials, we hypothesized that BGG492 is effective in acute migraine attacks. We conducted the first proof-of-concept (PoC) Phase II study of BGG492 in subjects with acute migraine to determine whether BGG492 is efficacious in improving headache in migraine attacks.
Materials and methods
Study design
This was a multicenter, randomized, double-blind, parallel-group, active- and placebo-controlled PoC study conducted between May 2009 and August 2010 in subjects affected by acute migraine with or without aura (NCT00892203). A total of 75 subjects were randomized (1:1:1) to receive one single oral dose of either BGG492 (250 mg), sumatriptan (100 mg) or placebo. The selection of a 250 mg dose of BGG492 was near to the maximum-tolerated dose, which would enable detection of treatment effects in a PoC study, and was based on the safety and tolerability results of a single-dose study (data on file by Novartis). In addition, at 250 mg, only mild central adverse events (AEs) were reported, while cardiovascular events were more common at doses above 250 mg. Although no formal study comparing intravenous vs oral forms has been performed, the absolute bioavailability of BGG492 is estimated to be approximately 60–70% based on data from a radiotracer study (data on file by Novartis). Novartis Drug Supply Management used a validated system to generate randomization numbers and automate the random assignment of subjects to treatment arms in the specified ratio to ensure blinding. Subjects meeting eligibility criteria were assigned the lowest-available randomization number on the randomization list; this was recorded by the investigator on the electronic case report form. The randomization scheme for subjects was reviewed and approved by an internal biostatistics quality assurance group.
All subjects completed a screening period of up to 28 days at one of 14 study centers in Germany, Spain and the United States that were qualified for diagnosis and treatment of migraine. Treatment occurred on the day of the first symptom of a migraine attack following the screening visit (within 28 days of screening). Subjects were required to report to the study center within four hours of onset. Attacks had to be moderate or severe, with or without aura. Upon reaching the center, baseline assessments were performed, and if appropriate, the subject was randomized and administered one single dose of the randomized treatment. All treatments were identical in appearance (gelatin capsules) to preserve blinding and were administered orally with 240 ml of water. All subjects, investigator staff, and persons performing the assessments were blinded for the duration of the study. Contrary to the initial study protocol, the clinical trial leader remained unblinded throughout, but had no contact with study subjects. The statistician, programmer, and translational medicine expert were unblinded at the time of interim analysis (details of interim analysis below).
After dosing, subjects were required to remain in the center for 6 hours to undergo efficacy and safety assessments. If no headache improvement was detected after 4 hours post-dosing (or earlier if deemed necessary by the investigator), subjects were treated with an appropriate rescue medication (paracetamol, acetylsalicylic acid, or ibuprofen). After completion of all scheduled assessments, subjects were required to attend a study completion visit 2 ( ± 1) days post-drug administration.
Study population
The study protocol was reviewed by the independent ethics committee or institutional review board at each study center. The study was conducted according to the ethical principles of the Declaration of Helsinki. All subjects were required to provide informed written consent before randomization. The authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of data analysis.
The study included healthy male and female subjects aged 18–60 years. Subjects had a diagnosis of migraine and had experienced migraine episodes of moderate or severe intensity, with or without aura (International Headache Society (IHS) categories 1.1 and 1.2) for at least one year prior to study entry. Subjects were required to have experienced more than one migraine episode, but no more than 15 migraine days per month, for each of the 6 months prior to study start. Migraine onset before the age of 50 years and prior history of triptan treatment were also required.
Subjects were excluded if they had: a diagnosis of basilar-type, retinal (ophthalmoplegic) or hemiplegic migraine; non-migraine headaches on more than 6 days per month over the previous 6 months; used any treatment for migraine attack prior to study drug administration; received regular treatment with psychoactive drugs in the 4 weeks prior to study start; used migraine prophylactic treatments or medications that interact with cytochrome P450 3A4 or P-glycoprotein in the 4 weeks before initial dosing; used medications contraindicated for sumatriptan within 1 week before initial dosing; any psychiatric condition that may put the subject at risk or affect efficacy assessments; used tobacco products in the preceding 3 months.
Objectives
The primary objectives were to assess the efficacy of a single dose of BGG492 vs placebo and an active comparator (sumatriptan) in subjects with acute migraine using the Patient Migraine Diary, and to evaluate the safety and tolerability of BGG492 by monitoring side effects. The secondary objective was to use the Patient Migraine Diary to evaluate the improvement in pain score, sustained response rate, pain-free rate, and sustained pain-free rate. Further exploratory objectives were to determine the pharmacokinetic (PK) profile of BGG492 in subjects with acute migraine and explore the PK/pharmacodynamic (PD) relationship.
Only the primary endpoint was stated on ClinicalTrials.gov, in accordance with Novartis policy. The primary objective, along with all other objectives and endpoints, was pre-specified in the study protocol (release date: 30 September 2008) and there were no related protocol amendments.
Efficacy assessments
Primary and secondary efficacy outcomes were assessed using the Patient Migraine Diary, a self-assessment questionnaire that rates the severity of five symptoms (headache, nausea, vomiting, photophobia, and phonophobia). Each symptom was rated on a four-point severity scale ranging from none (grade 0) to severe (grade 3). Efficacy was assessed on Day 1 at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, and 24 hours post-dose. The primary outcome measure was the percentage (%) of subjects with an improvement in headache from severe or moderate to mild or no pain and was measured at 2, 3, and 4 hours post-dosing (with no rescue medication before the relative timepoint). Other assessments included: (i) ≥ 2-point improvement in pain score (% of subjects with 2-point improvement in pain score from baseline, and no rescue medication before the relative timepoint); (ii) pain-free rate (% of subjects with no headache at 2, 3, and 4 hours post-dosing, and no rescue medication before the relative timepoint); (iii) sustained response rate (% of responders without rescue or recurrence from 4 to 24 hours); (iv) sustained pain-free rate (% of pain-free subjects without rescue or recurrence from 4 to 24 hours); (v) recurrence rate (% of responders who experience a grade 2/3 headache on the 4-point severity scale from 4 to 24 hours); and (vi) non-pain measures such as relief of nausea, photophobia, or phonophobia at 2, 3 or 4 hours, in all evaluable subjects.
PK and PD assessments
Blood samples were collected at 0, 0.25, 0.75, 1.0, 1.5, 2, 3, 4, 5, and 6 hours post-dose. Assessed PK parameters included: the area under the curve from drug administration to 2 hours (AUC0–2h); the AUC from drug administration to the last measureable concentration sampling time (AUClast); the maximum plasma/blood/serum concentration after single dose administration (Cmax); lag-time (Tlag); the time to reach Cmax (Tmax); and the elimination half-life associated with the terminal slope (lambda z) of a semi-logarithmic concentration-time curve (T1/2).
Safety
All AEs and serious AEs (SAEs) with their severity and relationship to the study drug were recorded. During the treatment period hematology, blood chemistry, and urine samples were monitored, and regular assessments were taken of vital signs, physical condition, and body weight. Females were required to have negative pregnancy test results at screening and at baseline and were required to use two acceptable methods of contraception up to 30 days following study completion.
Statistical analysis
Determination of sample size was based on the primary outcome measure of the study and simulations, taking into account the interim analysis, the final analysis, and the dependence between responses at the three timepoints. It was planned that 75 subjects would be recruited; with evaluable 25 subjects in each treatment arm, it was calculated that if the difference between the true response rates (of BGG492 and placebo) was greater than 35%, the probability that the trial would pass the PoC rule was ≥ 0.86. If there was no significant difference between the true response rates (< 10%), the probability that the trial would pass the PoC rule was lower than 0.05.
For efficacy measures, analysis with a generalized linear model (logit link function) was conducted 2, 3, and 4 hours post-dose with treatment and time as factors. Estimates and 90% confidence intervals were determined for the response rate of each treatment at each timepoint. PoC was regarded as established if (i) at interim analysis (when 15 subjects in each treatment group completed the study), the difference between the response rates of BGG492 and placebo was ≥ 35% in at least two of the specified timepoints (2, 3, and 4 hours post-dose), or (ii) at final analysis, the difference was ≥ 25% in at least two of the specified timepoints. If PoC had been met at interim analysis, the study would have been stopped.
Descriptive statistics of PK parameters included mean, standard deviation (SD), and % coefficient of variation (% CV; min and max). Overall and % CV geometric means are also presented. Since Tmax is generally evaluated by a non-parametric method, median values and ranges were given for this parameter. Concentrations below the limit of quantification were treated as zero in summary statistics of concentration data and for calculation of PK parameters. The relationship between BGG492 response rate (PD) and PK parameters was analyzed by a generalized linear model (logit link function) with time and PK parameter as explanatory variables. The threshold for statistical significance compared with placebo was p < 0.05.
The safety cohort included all subjects who received the study drug. Subjects were analyzed according to treatment received. The PK cohort included all subjects in the safety cohort with evaluable PK parameter data and with no major protocol deviations that may have influenced PK data. The PD cohort included all subjects in the safety cohort with both pre-dose and post-dose PD measurements (at least at 2, 3, and 4 hours post-dose) and with no major protocol deviations that may have influenced PD data.
Results
Study population
A total of 75 subjects were enrolled and randomized in the study and all completed with no discontinuations. All 75 subjects were included in the safety cohort with 25 subjects in each treatment arm (Figure 1). A major protocol deviation was reported by one subject in the BGG492 group; the severity of migraine was deemed to be mild at a pre-dose timepoint. This subject was randomized in error and would have been counted as a “responder” according to the primary outcome (improvement to mild or no pain). This would not have been a true treatment effect and so the subject was excluded from PD analysis (total of 74). All other protocol deviations were considered as minor. A total of 50 subjects were included in the PK cohort (N = 25 in the BGG492 group and N = 25 in the sumatriptan group).
Flow of subjects through the study.
Demographics
Subject demographics (safety cohort).

BMI: body mass index; N: total number treated; n: number in group; PD: pharmacodynamic. aPresented as mean (standard deviation).
In the PD cohort (N = 25 in sumatriptan, N = 24 in BGG492, and N = 25 in placebo).
Primary efficacy outcome
In the interim analysis, at 2, 3 and 4 hours post-dose, the response rate (defined as headache improvement from severe or moderate to mild or no pain, with no rescue medication) for BGG492 was 56.3% (9/16) at all timepoints, compared with 39.1% (9/23), 47.8% (11/23), and 43.5% (10/23) for placebo, respectively. In the final analysis at 2 hours post-dose, response rates were 68% (17/25) for sumatriptan, 58% (14/24) for BGG492, and 40% (10/25) for placebo (Figure 2). Responses at 3 and 4 hours are presented in Table 2; despite the higher number of responders in the BGG492 group compared with placebo, BGG492 was not significantly better than placebo and the PoC criterion for efficacy was not met as the number of responders did not differ by ≥ 25% at two of the specified timepoints (Table 2).
Primary endpoints at 2 hours, as measured by the Migraine Patient Diary (PD cohort). Improvement in pain as evaluated by the Patient Migraine Diary (PD cohort). PD: pharmacodynamic; N: number analyzed; CI: confidence interval; LL: lower limit; UL: upper limit. aCompared with placebo. Data presented as number of subjects and percentage of total in treatment group. Numbers include those that did not receive rescue medication before the given timepoint. ≥ 2-point improvement defined as an improvement from baseline of two or more on the four-point severity scale.

Other efficacy outcomes
Other PD variables included a ≥ 2-point improvement in pain score and pain-free response without rescue medication before the given timepoint (Figure 2); according to these measures, BGG492 was significantly better than placebo at 4 hours (Table 2). At 2 hours post-dose, 25% (6/24) of subjects in the BGG492 group reported a pain-free response compared with 16% (4/25) in the placebo group and 24% (6/25) in the sumatriptan group; BGG492 was significantly better than placebo at 4 hours post-dose (Table 2 and Figure 2).
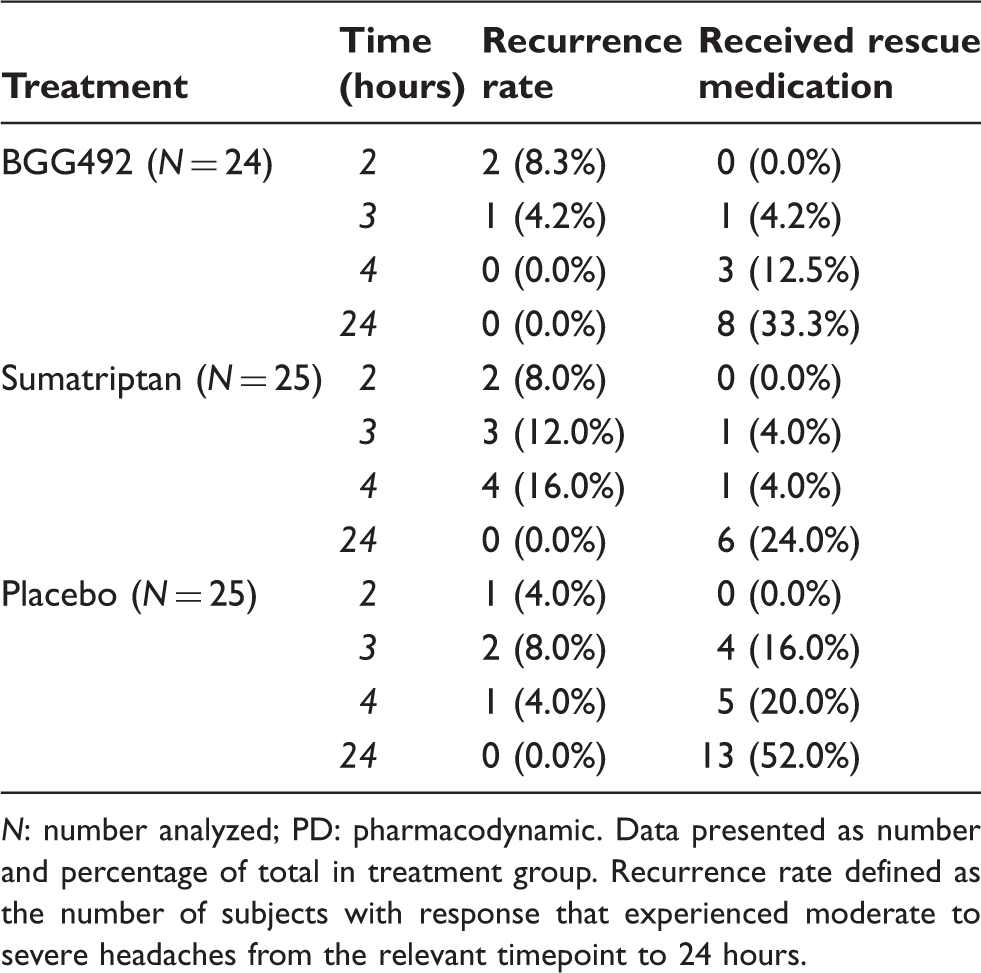
Sustained response rate, sustained pain-free rate, and recurrence assessments indicate that a sustained response to BGG492 was observed at 24 hours in some subjects (Figure 3 and Table 3). Responses were maintained to 24 hours in 54% (13/24) of BGG492 and 36% (9/25) of placebo subjects who reported a response at 4 hours (Figure 3). Recurrence of headaches in the BGG492 group occurred at 24 hours in 8% (2/24) and 4% (1/24) of subjects who reported a response at 2 and 3 hours, respectively. No recurrences were reported among subjects with a response at hour 4. Recurrences for sumatriptan and placebo groups are given in Table 3. In total, 27 subjects received rescue medication by the 24-hour timepoint (Table 3).
Sustained responses as evaluated by the Migraine Patient Diary (PD cohort). Sustained response rate or pain-free response includes subjects that reported a headache improvement to mild or no pain or pain-free response, that persisted from the relevant timepoint through to 24 hours without rescue medication or recurrence. Rates of recurrence and administration of rescue medication through 24 hours as evaluated by the Patient Migraine Diary (PD cohort). N: number analyzed; PD: pharmacodynamic. Data presented as number and percentage of total in treatment group. Recurrence rate defined as the number of subjects with response that experienced moderate to severe headaches from the relevant timepoint to 24 hours.
There were more subjects in the sumatriptan group (23/25) than the BGG492 group (16/24) who demonstrated an improvement to mild or no pain by at least one of the timepoints (2, 3, and 4 hours). However, the mean time to onset of response was shorter in the BGG492 group at 1.1 hours compared with the sumatriptan group (1.7 hours). In the placebo group, 48% (12/25) of subjects responded with a mean time of onset of 1.4 hours.
Scores for non-pain measures decreased overall for all treatment groups, except for vomiting in the sumatriptan group up to 4 hours post-dose. Scores decreased by 0.1–0.2 points for vomiting, 0.2–0.5 for nausea, 0.6–1.2 for photophobia, and 0.6–1.0 for phonophobia, and were generally numerically similar between treatment groups.
PK analysis
The dose of BGG492 used was high, thus, biologically relevant concentrations were attained shortly after dosing. Both drugs were measurable in the plasma as early as 0.25 hours post-dose following a single dose (250 mg BGG492 or 100 mg sumatriptan; Figure 4 and Table 4). The inter-subject variability in both groups was moderate in terms of Cmax and AUClast; however, the % CV geometric mean of AUC0–2h was 464% for BGG492 compared with 36% for sumatriptan (Table 4). Although Cmax and Tmax were variable, concentrations of BGG492 generally increased quickly after dosing and then reached a broad plateau, which could account for the high variability.
Concentration time profile following single oral administration of BGG492 or sumatriptan in the PK cohort. Plasma PK parameters following single oral administration of BGG492 or sumatriptan (PK cohort). PK: pharmacokinetic; N: number analyzed in group; SD: standard deviation; CV%: percentage coefficient of variation; Geo-mean: geometric mean; AUClast: area-under-curve from time zero to the last measurable concentration sampling time (tlast) [mass × time × volume]; AUC0–2h: AUC from time zero to 2 hours [mass × time × volume]; Cmax: peak observed plasma concentration; Tmax: time to reach maximum plasma concentration; Tlag: lag-time. aStated as a percentage.

A PK/PD model was developed for both BGG492 and sumatriptan to assess concentration effects according to the three responder endpoints: improvement to mild or no pain; ≥ 2-point improvement; and pain-free response. The effects of BGG492 concentration on the responder endpoints were not statistically significant. However, the concentration effects of sumatriptan were significant for the ≥ 2-point improvement endpoint.
Tolerability and safety
Reported adverse events by body system (safety cohort).
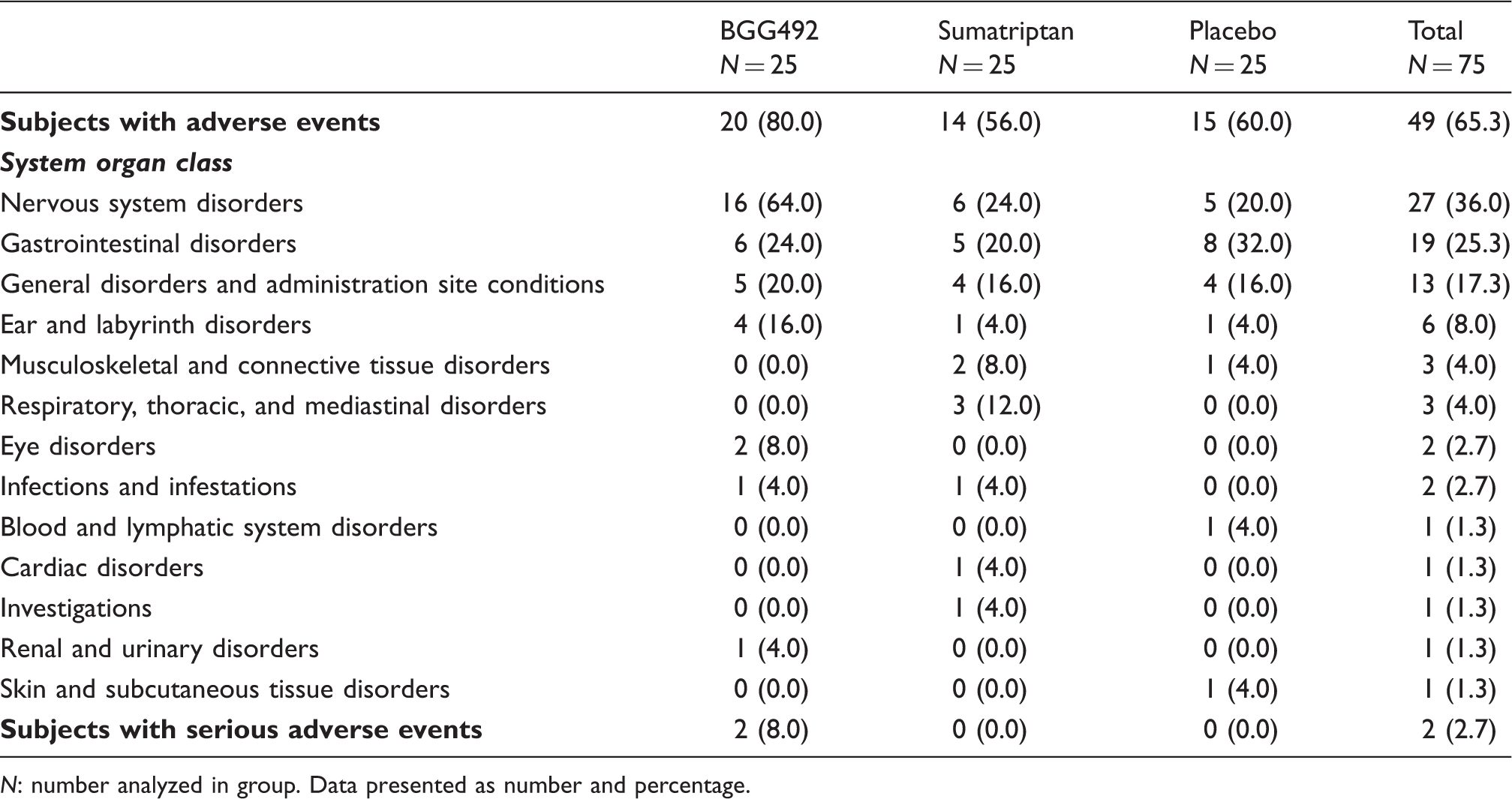
N: number analyzed in group. Data presented as number and percentage.
Discussion
The objective of this PoC study was to determine whether BGG492, compared with placebo and sumatriptan, is efficacious in suppressing acute migraine pain. While BGG492 treatment did result in improvement of headache pain and pain-free response in a subset of subjects, the primary PoC criterion was not met as the difference between the number of responders to BGG492 and placebo was not ≥ 25% at two timepoints. It is noteworthy that the pain-free responses observed with BGG492 at all timepoints, including the IHS preferred outcome of pain-free at 2 hours (10), were similar to sumatriptan and a sustained response was seen at 24 hours with BGG492 also. Along with these positive effects, a relatively high response rate was observed in the placebo group.
High placebo responses are often observed when migraine patients are potentially exposed to a new migraine drug as this leads to a high treatment effect expectation. In addition, subjects were treated in-house, which may have contributed to the placebo effect and also limits comparisons with other oral triptan trials. In-house treatment also led to a delay in treatment following the onset of migraine attack, which may have resulted in more intense head pain, further limiting the comparisons with other trials. However, conducting treatment and observation in headache centers also adds strength to the stringent protocol. Other limitations include the small sample size with insufficient power for the endpoint proposed by the IHS (10) and the fact that patients may have been unblinded because of the side effects of BGG492.
Although BGG492 did not meet the PoC criterion, it did provide migraine pain relief in some subjects. This supports the idea that AMPA receptor antagonists are potentially efficacious as anti-migraine drugs. One possible mode of action is a decrease in excitability and subsequent sensitization, brought about by inhibition of glutamatergic transmission in the TNC. AMPA receptor antagonists may also be implicated in the earlier stages of migraine pathophysiology. CSD, which is thought to underlie the aura that some migraine patients experience prior to onset of pain, relies heavily on glutamatergic transmission; in animal models, CSD-associated blood flow changes were inhibited by an AMPA antagonist (11). Thus, in humans, blockade of AMPA receptors may affect a possible triggering mechanism of headache. In contrast, it has been suggested that activation of AMPA receptors may suppress the actions of NMDA receptors, which also play a crucial role in CSD; in this scenario, AMPA antagonists would increase NMDA activity and so could increase CSD (12). Further studies are required, using a study design that allows earlier administration of treatment, to investigate this and especially to address the potential effect on aura symptoms. In this study, time of administration may have affected the outcome as this was performed in-clinic, which probably delayed the treatment effect. Thus, further investigation into the effects of early administration on BGG492 efficacy is required.
Another aim of this study was to investigate the PK characteristics of BGG492 in subjects with acute migraine. In general, the findings were similar to those in a previous study involving healthy volunteers receiving the same concentration of BGG492 (Novartis, data on file). However, the inter-subject variability in AUC0–2h was considerably higher in this study; the % CV geometric mean value was 464% in subjects experiencing acute migraine, compared with 77% in healthy subjects (Novartis, data on file), which suggests that the disease state may affect initial absorption of BGG492. For sumatriptan in the encapsulated form, the PK data here were consistent with those reported in a previously published study (13).
The AEs observed in this study in the BGG492 group were consistent with the known safety profile of the drug. Only two subjects in the BGG492 group experienced SAEs, which were mild in severity and resolved without intervention. However, the incidence of AEs affecting the central nervous system (CNS) in the BGG492 group was quite high. Nearly half of the subjects in the BGG492 group experienced dizziness, which would have prevented some from performing daily activities; this would therefore be a limiting factor when considering the benefits of this compound vs AEs. Further studies would be required to fully elucidate the safety profile of BGG492, particularly with regards to the potential for CNS AEs. In addition, no conclusions can be made regarding the safety profile of BGG492 following repeat dosing.
The PoC criterion in this study was not met. BGG492 provided pain relief in only a subset of subjects with acute migraine, and many subjects experienced AEs. Further investigations are warranted to determine the characteristics of the subjects who experienced headache relief with BGG492 (e.g. attack severity, frequency, and duration and presence of allodynia and/or aura), and to fully elucidate the efficacy of glutamate receptor antagonists in migraine therapy. Factors to consider when designing future trials include the optimum strategy with regards to time of administration.
Clinical implications
In this randomized, double-blind trial, efficacy of a novel amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor antagonist, BGG492, was compared with placebo and sumatriptan, in subjects with acute migraine, as assessed using the Patient Migraine Diary in specialized headache centers. Although improvement from severe or moderate to mild or no headache pain (primary endpoint) was reported in a subset of subjects receiving BGG492, the proof-of-concept criterion was not met. BGG492 was comparable to sumatriptan in terms of pain-free and sustained responses. Further investigations are required to determine the characteristics of subjects who may benefit from BGG492 treatment in migraine.
Footnotes
Funding
This study was funded by Novartis Pharma AG, Basel, Switzerland, and is registered at clinicaltrials.gov (NCT00892203).
Conflicts of interest
Ronald Brand has received honoraria for clinical studies from Boehringer Ingelheim.
Tim Jürgens has received honoraria for talks from MSD Germany and Autonomic Technologies Inc. and serves as a consultant for Autonomic Technologies Inc.
Harmut Göbel has advised, collaborated with, and spoken at meetings organized by companies including: Allergan, Almirall-Prodesfarma, AstraZeneca, Bayer Vital, Berlin-Chemie, Bionorica, Bristol-Myers Squibb, Fujisawa, GlaxoSmithKline, Grünenthal, Hermal, Ipsen-Pharma, Janssen-Cilag, Johnson & Johnson, Krewel-Meuselbach, Lichtwer, Menarini Pharma, Merz Pharmaceuticals, Minster Pharmaceuticals, MSD, Novartis, Pfizer, Pharmacia, Sandoz, Schaper und Brümmer, Schwarz-Pharma, Weber & Weber, and SmithKlineBeecham.
Claudia Sommer has given educational talks for Allergan Inc. and GlaxoSmithKline.
Andreas Straube has received honoraria for educational lectures from: Pfizer, Allergan, MSD, Böhringer Ingelheim, is/was a member of advisory boards for MSD and Allergan, and has received financial support from the German science council (DFG), the federal secretary for education (BMBF), and the University of Munich.
Stefan Evers has received honoraria for participation in clinical trials and contributions to advisory boards or oral presentations from: Addex Pharma, Allergan, Almirall, AstraZeneca, Berlin Chemie, Böhringer Ingelheim, Eisai, GlaxoSmithKline, Ipsen, Janssen-Cilag, Merz, MSD, Novartis, Pierre Fabre, Pfizer, sanofi aventis, UCB, and Weber & Weber.
Martin Sommer's institution received fees for the study presented here (Novartis). MS has received honoraria for educational lectures from GlaxoSmithKline, Lundbeck, and Licher Medizintechnik.
Victor Campos has received honoraria for educational lectures from GlaxoSmithKline, Lundbeck, Novartis, Böhringer Ingelheim, Janssen-Cilag, Allergan, MSD, UCB pharma, and Abbott.
Hans-Christoph Diener has received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from: Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, Böhringer Ingelheim, Bristol-Myers Squibb, Coherex, CoLucid, GlaxoSmithKline, Grünenthal, Janssen-Cilag, Lilly, La Roche, 3M Medica, Medtronic, Menerini, Minster, MSD, Neuroscore, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brümmer, Sanofi, St. Jude, and Weber & Weber. Financial support for research projects was provided by: Allergan, Almirall, AstraZeneca, Bayer, GSK, Janssen-Cilag, MSD, Novartis, and Pfizer. Headache research at the Department of Neurology in Essen is supported by the German Research Council (DFG), the German Ministry of Education and Research (BMBF), and the European Union. Hans-Cristoph Diener has no ownership interest and does not own stocks of any pharmaceutical company.
Hans O Kalkman, Sam Hariry, Nicole Pezous, Donald Johns, and Baltazar Gomez-Mancilla are employees of Novartis Pharma AG.
Acknowledgment
Medical writing and editorial assistance in the development of this manuscript were provided by Louisa Pettinger, Fishawack Communications Ltd (Cheshire, UK), supported by Novartis Pharma AG, Basel, Switzerland.