
Abstract
Background
Familial hemiplegic migraine type 1 (FHM-1) is an autosomal dominant form of migraine with aura characterized by recurrent migraine, hemiparesis and ataxia. FHM-1 has been linked to missense mutations in the CACNA1A gene encoding the pore-forming subunit of the neuronal voltage-gated P/Q-type Ca2+ channel (CaV2.1α1).
Methods
Here, we explored the effects of the FHM-1 K1336E mutation on G protein-dependent modulation of the recombinant P/Q-type channel. The mutation was introduced into the human CaV2.1α1 subunit and its functional consequences investigated after heterologous expression in HEK-293 cells using patch-clamp recordings.
Results
Functional analysis of the K1336E mutation revealed a reduction of Ca2+ current densities, a ∼10 mV left-shift in the current-voltage relationship, and the slowing of current inactivation kinetics. When co-expressed along with the human μ-opioid receptor, application of the agonist DAMGO inhibited whole-cell currents through both the wild-type and the mutant channels. Prepulse facilitation was also reduced by the K1336E mutation. Likewise, the kinetic analysis of the onset and decay of facilitation showed that the mutation affects the apparent dissociation and reassociation rates of the Gβγ dimer from the channel complex.
Conclusions
These results suggest that the extent of G-protein-mediated inhibition is significantly reduced in the K1336E mutant CaV2.1 Ca2+ channels. This alteration would contribute to render the neuronal network hyperexcitable, possibly as a consequence of reduced presynaptic inhibition, and may help to explain some aspects of the FHM-1 pathophysiology.
Introduction
Migraine is a chronic neurological disorder with recurrent headache episodes. This condition affects ∼20% of individuals in the general population and has been classified in two major types: 1) migraine without aura occurring in about two-thirds of patients, which is characterized by attacks of unilateral pulsating headache usually associated with nausea and photo- and phono-phobia, and 2) migraine with aura in which the headache episodes are preceded by transient focal neurological symptoms (1–4). Clinical studies have shown that many patients have first-degree relatives who also suffer from the disease (5), and therefore its inherited nature is an interesting aspect of the migraine pathophysiology (4).
Familial hemiplegic migraine (FHM) falls within the category of migraine with aura. In FHM the neurologic symptoms are localizable to the cerebral cortex or brain stem and include visual disturbance, sensory loss, dysphasia and hemiparesis. Three genes are known to be associated with FHM. Mutations in CACNA1A and SCNA1A, encoding the ion-conducting subunits of the neuronal voltage-gated P/Q-type Ca2+ (CaV2.1α1) and Na+ (NaV1.1) channels, are responsible for FHM types 1 and 3 (FHM-1, FHM-3), respectively, whereas mutations in ATP1A2, coding the α2 subunit of the Na+–K+ ATPase, are responsible for FHM-2 (4,6,7). It should be noted that migraine is a complex genetic disorder. Recent genome-wide association studies have identified a few risk factors for migraine that map within or near transcribed regions of interesting genes including metadherin (MTDH), a gene that regulates the expression of GLT-1, an astrocyte glutamate transporter that plays a major role in removal of glutamate at glutamatergic synapses, and transient receptor potential melastatin 8 (TRPM8), a gene that encodes a cation channel expressed in sensory neurons (8–10).
Functional consequences of FHM-1 gene mutations have been studied in cellular and animal models. At least 13 of the 21 reported FHM-1 mutations have been tested in heterologous expression systems for their consequences on Ca2+ channel function using electrophysiology (4,6,7). These studies have shown that FHM-1 mutations alter different properties of human CaV2.1 channels. For instance, an enhanced-channel open probability and single-channel Ca2+ influx associated with a shift to lower voltages of channel activation has been reported (11,12). Likewise, using an FHM1 transgenic mouse model, it has been shown that the FHM-1 R192Q mutation produces gain-of-function (13–17). More recently, studies performed on cortical slices from FHM-1 knock-in mice have shown that diverse synapses (excitatory and inhibitory) may be differentially affected by FHM-1 mutations (10,14).
On the other hand, though decreased Ca2+ current densities have also been found for other FHM-1 mutants (11,12,18,19) expressed in heterologous systems, investigation of synaptic transmission in CaV2.1α1 null neurons transfected with FHM-1 mutant CaV2.1 channels revealed an unaltered overall synaptic strength (20,21). Hence, based on the available data, it is generally accepted that FHM-1 mutations lead to gain of function, which facilitates the propagation of cortical spreading depression (CSD) (13,22) as a consequence of increased glutamate release from cortical pyramidal cell synapses (14). CSD is considered a key event underlying migraine aura (23).
Much less is known regarding the effects of FHM-1 mutations on the modulation of CaV2.1 channels by G proteins. Although no FHM-1 mutations have been found in the channel region involved in G-protein binding, various studies revealed a reduction of G-protein-mediated channel inhibition (24–27), an effect that may lead to altered Ca2+ influx through mutant channels during neuromodulation. Here, we studied the unknown functional consequences of an additional FHM-1 mutation (K1336E) located in the third repeat domain of the CaV2.1α1 subunit on G-protein-mediated inhibition. Our results show that the extent of G-protein-mediated inhibition and prepulse facilitation are significantly reduced in the mutant recombinant P/Q-type Ca2+ channels, thereby supporting the pathogenicity of this FHM-1 mutation. These results are consist with the hypothesis that FHM-1 mutations produce gain-of-function of human recombinant CaV2.1 channels, since neuronal hyperactivity may also be caused by a reduction of the inhibitory pathway carried by G-protein-coupled-receptor (GPCR) activation.
Materials and methods
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells were maintained in Dulbecco’s modified essential medium (DMEM)-high glucose supplemented with 10% horse serum, 1% L-glutamine, 110 mg/l sodium pyruvate and antibiotics, at 37°C in a 5% CO2–95% air humidified atmosphere. Gene transfer was performed using Lipofectamine Plus reagent as described previously (27). Briefly, for a 35-mm Petri dish of HEK-293 cells, 2 µg of the plasmid cDNA encoding the wild-type (WT) or the K1336E mutant of the human P/Q-type Ca2+ channel CaV2.1α1 pore-forming subunit splice isoform 1 (GenBank accession number AF004883), in combination with 2 µg CaVβ3 (M88751) and 2 µg of CaVα2δ-1 b cDNA (M86621), were mixed with 6 μl of Lipofectamine in 100 μl serum-free medium according to the manufacturer’s instructions. The solution was then added to the dish and cells grown at 37°C for 24 hours, when medium was changed.
FHM-1 K1336E mutation was introduced into the cDNA coding the CaV2.1α1 as previously reported (27). Mutant protein-coupled receptors products harboring the mutation were subcloned into a mammalian expression vector, and the presence of the mutation was verified by automated DNA sequencing (Supplementary Figure 1(a)). The cDNA coding the human µ-opioid receptor (hMOR; AY521028) was obtained from the University of Missouri-Rolla (UMR) cDNA Resource Center (www.cdna.org) and used as described elsewhere (25,27). Briefly, the hMOR cDNA was inserted into the pcDNA3.1 vector and transfected along with the Ca2+ channel subunits into HEK-293 cells by using Lipofectamine. Transfections were performed with 2 µg of DNA plasmid and 6 μl of Lipofectamine in DMEM-reduced serum medium in 35-mm plates. Functional studies were performed 48 hours posttransfection.
Electrophysiological recoding
Ba2+ currents (IBa) through recombinant Ca2+ channels were recorded using the whole-cell configuration of the patch-clamp technique (28). The extracellular solution contained (in mM): BaCl2, 10; tetra-ethylammonium chloride (TEA-Cl), 125; 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 10; glucose, 10 (pH 7.3). The pipette solution contained (in mM): CsCl, 110; MgCl2, 5; ethylene glycol tetraacetic acid (EGTA), 10; HEPES, 10; adenosine 5'-triphsophate (ATP) disodium salt, 4; guanosine-5'-triphosphate (GTP), 0.1 (pH 7.3). Recordings were performed using an Axopatch 200B amplifier, and currents were digitized at a sampling rate of 5.7 kHz. Linear components were subtracted online using a P/4 protocol. Membrane capacitance (Cm) was determined as previously described (29) and used to normalize currents.
Current-voltage (I-V) relationships were obtained by step depolarization between −50 mV and +70 mV in 10 mV increments, from a holding potential (Vh) of −80 mV. Steady-state inactivation was studied by applying 10 s conditioning pulses to various potentials before a test depolarization at +10 mV. Inactivation curves were fitted with Boltzmann equations. Time constant of channel inactivation was obtained from single exponential fits of IBa decaying phase using 250-ms test pulse to +10 mV. The voltage-dependence of IBa inhibition by G-proteins was analyzed by applying a series of 25-ms pulses to various voltages (ranging from −50 mV to +70 mV in 10 mV steps) 500 ms after (P1) and 5 ms before (P2) a 50-ms conditioning prepulse to +100 mV, from a Vh of −80 mV. Facilitation was then determined as the ratio IBa (P2/P1). Facilitation decay and development was studied as previously reported (24,26,27), applying a three-pulse voltage protocol (Supplementary Figure 1(b)). Plots of IBa (P2/P1) as a function of time were fitted to single exponentials in order to obtain the corresponding time constants. In all cases, the number of recorded cells is given in the corresponding figure legends.
Structural modeling of repeated domain III in the CaV2.1α1 subunit
Homology models of domain III (residues 1243–1509) in the human CaV2.1α1 subunit isoform 1 were generated using the Robetta server (http://robetta.bakerlab.org). The WT sodium channel from Arcobacter butzleri (NaVAb) inactivated-state model (30) was used as a template. The backbone conformation of the models generated was validated using the Structural Analysis and Verification Server (http://nihserver.mbi.ucla.edu/SAVES/Info.php). The best scored structure obtained from the Robetta server was chosen as a template to introduce the K1336E mutation using the University of California, San Francisco (UCSF) Chimera software (http://www.cgl.ucsf.edu/chimera/). The position of the mutated residue was optimized to regularize local bond and angle geometry and remove bad contacts. Finally, the two structure models of domain III (WT and mutated) were oriented in a lipid bilayer using the Positioning of Proteins in Membranes (PPM) server (http://opm.phar.umich.edu/server.php) (31) and the electrostatic potential calculated using the Swiss-Pdb Viewer software (http://www.expasy.org/spdbv/).
Results
FHM-1 has been linked to missense mutations in the CACNA1A gene encoding the CaV2.1α1 ion-conducting subunit of the neuronal P/Q-type Ca2+ channels. CaV2.1α1 consists of four repeated domains (I–IV), each containing six transmembrane regions (S1–S6) with a voltage sensor (S1–S4) and a pore loop between S5 and S6. The mutation studied here (K1336E) is located in the loop region connecting segments S3 and S4 of repeated domain III (Figure 1(a)).
K1336E mutation affects P/Q-type current properties and channel expression. (a) Location of the K1336E mutation in the secondary structure of the CaV2.1α1 subunit (upper panel). Representative recordings illustrating the amplitude and kinetics of current through wild-type (WT) and K1336E channels in response to depolarizing pulses to +10 mV from a holding potential (Vh) of −80 mV. (b) Current density-voltage relationships for WT and mutant channels expressed in HEK-293 cells (n = 7–8). (c) Steady-state inactivation curves of currents through WT or mutant channels as indicated. Currents were elicited by 10 s conditioning pulse from a Vh of −80 mV in 10 mV steps from −110 to +20 mV followed by a test pulse to +10 mV. The individual data points are means of five to nine recorded cells. Error bars reflect standard errors, and the solid lines reflect fits via a Boltzmann function. V½ values were −43.89 mV and −47.55 for WT and FHM-1 mutant K1336E channels, respectively. (d) Comparison of time constant of inactivation in cells expressing WT and FHM-1 mutant channels (n = 10–12; *p < 0.05).
We first characterized the impact of the K1336E mutation on current density by transiently expressing CaV2.1α1 WT and K1336E variants (together with CaVβ3 and CaVα2δ-1 b channel auxiliary subunits) in HEK-293 cells and recording whole-cell patch clamp currents two days after transfection (Figure 1(a)). As can be seen in Figure 1(b), current density recorded was significantly decreased in the FHM-1 mutant K1336E in comparison with the current density recorded in WT CaV2.1 channels at +10 mV (−22.6 ± 3.4 and −40.7 ± 3.5 pA/pF, respectively). The potential for half-maximal activation was significantly shifted (∼10 mV) in the hyperpolarizing direction for the mutant K1336E channels without apparent changes in the reversal potential. Consistently, the maximum current amplitude was evoked by depolarizing pulses to +10 or 0 mV for the WT or the mutant channels, respectively (Figure 1(b)). On the other hand, the half-maximal voltage for steady-state inactivation induced by 10 s conditioning prepulses ranging from −110 to +50 mV was slightly left-shifted (∼5 mV) in the K1336E channels (Figure 1(d)), and such an effect was accompanied by a significant slowdown in IBa decay during 250 ms test pulses evoked from a Vh of −80 mV to +10 mV (Figure 1(d)).
Given that leftward shifts in voltage-dependence of activation and slowed inactivation rate have been reported to facilitate recovery from direct G protein regulation (25), we next investigated the effects of the K1336E mutation on voltage-dependent Gβγ-mediated inhibition of recombinant P/Q-type channels. To this end, current inhibition by the μ-opioid receptor (hMOR) was examined using [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), a synthetic agonist of hMOR. It is well known that hMOR couples to pertussis toxin (PTX)-sensitive Gi/Go G-proteins to inhibit Ca2+ channels (32).
IBa was recorded from HEK-293 cells transiently transfected with either the WT or the K1336E mutant channels together with hMOR. Exposure to DAMGO (10 μM) produced a rapid (within 10 s) voltage-dependent inhibition of the current through both WT and K1336E mutant channels (Figure 2(a)). The inhibition was most evident at the peak IBa (0 to +10 mV) in both WT and mutant channel expressing cells, and more effective in WT channels at depolarized potentials (Figure 2(b)). Interestingly, the K1336E mutation caused a significant reduction in the magnitude of such inhibition. DAMGO inhibited IBa ∼17% in both WT and K1336E channels expressing cells at −10 mV; in contrast at +10 mV DAMGO reduced IBa by ∼48% in the WT channel cells expressing and ∼36% in cells expressing K1336E mutant channels.
K1336E mutation alters receptor-mediated G-protein CaV2.1 channel regulation. (a) Representative current traces recorded from cells expressing wild-type (WT) and K1336E channels at +10 mV before and after [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) application showing the inhibition of the peak current under direct G-protein regulation. (b) Comparison of mean values for the peak current inhibition produced by DAMGO application as a function of test potential applied (n = 8–9 recorded cells; *p < 0.05). (c) Representative currents recorded during inhibition by 10 µm DAMGO before (P1) and after (P2) a 25 ms depolarizing prepulse to +100 mV. Test depolarizations to the indicated potentials were given 500 ms before and 5 ms after the prepulse. Currents are superposed to facilitate comparison. (d) Comparison of prepulse current facilitation recorded at +10 mV from cells expressing WT and K1336E channels. Currents after the prepulse were normalized to the maximal current amplitude before the prepulse and then averaged (n = 9–10 recorded cells; *p < 0.05).
G-protein modulation of CaV channels is characterized by its transient relief in response to a strong depolarizing prepulse. This is called facilitation and is due to the dissociation of Gβγ dimers as channels undergo conformational changes in response to depolarization. Therefore, we next investigated whether the K1336E mutation affected the recovery from G protein-mediated channel inhibition. Figure 2(c) shows current traces through WT (left panel) or mutant channels (right panel) recorded during inhibition by DAMGO and elicited by 25 ms test pulses to the indicated potentials before (P1) and after (P2) the prepulse (PP) to +100 mV. Co-expression of CaV2.1α1 WT or the K1336E mutation resulted in current facilitation, reflecting the voltage-dependent relief of Gβγ-mediated channel inhibition. Interestingly, a clear difference in the magnitude of facilitation was observed. The biggest reduction in facilitation was observed at +10 mV. At this potential, facilitation was significantly reduced from 1.47 ± 0.04 in the WT condition to 1.28 ± 0.05 in K1336E channels, respectively (Figure 2(d)).
These results, together with the observation that FHM-1 mutant channels are less sensitive to DAMGO inhibition, suggest either a lower level of Gβγ-mediated inhibition on the mutant channels or a facilitated deinhibition during the test pulse at higher voltages. Therefore, we sought to determine the mechanism underlying this difference by evaluating the association and dissociation rates of Gβγ dimers to/from the channel, which can be measured by varying the parameters of the three-pulse voltage protocol (Supplementary Figure 1(b)).
To monitor the onset of facilitation, the ratio of current amplitudes measured before and after the prepulse was plotted as a function of the conditioning prepulse duration (Figure 3(a) and 3(b)). For each cell, facilitation was fitted by a single exponential to obtain a time constant. As can be seen in Figure 3(c), facilitation developed with an averaged time constant of 5.04 ± 0.59 ms for WT and 3.44 ± 0.42 ms for K1336E mutant channels, indicating that the FHM-1 mutation promoted G protein dissociation from the channel during membrane depolarization. The decay of facilitation was monitored by plotting the ratio of current amplitudes measured before and after the prepulse as a function of a variable interval between the conditioning prepulse and the second test pulse (Figure 3(d) and 3(e)). For each cell, the decay of facilitation was fitted by a single exponential to obtain a time constant. Facilitation decayed with an averaged time constant of 27.7 ± 2.6 ms for WT and 19.8 ± 4.8 ms for K1336E channels (Figure 3(f)).
K1336E mutation affects the rate of G-protein association/dissociation from CaV2.1 channels. (a) Representative current traces recorded during relief of G-protein regulation induced by [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) (facilitation development) recorded from HEK-293 cells expressing wild-type (WT) and K1336E channels. Prepulse duration varied from 0 to 15 ms, and the interpulse intervals were fixed to 500 and 5 ms (Supplementary Figure 1(b)). Successive episodes of this voltage protocol were delivered at 20 s intervals. (b) Comparison of time course of facilitation development (P2/P1 ratio) as a function of facilitatory prepulse duration for WT and mutant channels. (c) Comparison of the time constant of G-protein dissociation from WT and K1336E mutant channels (n = 11–18 recorded cells; *p < 0.05). (d) Representative current traces recorded during re-inhibition by G-proteins (facilitation decay) of WT and K1336E mutant channels. The interval between the first test pulse (P1) and the prepulse (PP) was 500 ms; the interval (Δt) between PP and the second test pulse (P2) varied from 5 to 45 ms (Supplementary Figure 1(b)). Successive episodes of this voltage protocol were delivered at 10 s intervals. (e) Comparison of time course of facilitation decay (P2/P1 ratio) as a function of interpulse duration for WT and mutant channels. (f) Comparison of the time constant of G-protein reassociation from WT and K1336E mutant channels (n = 9–10 recorded cells; *p < 0.05).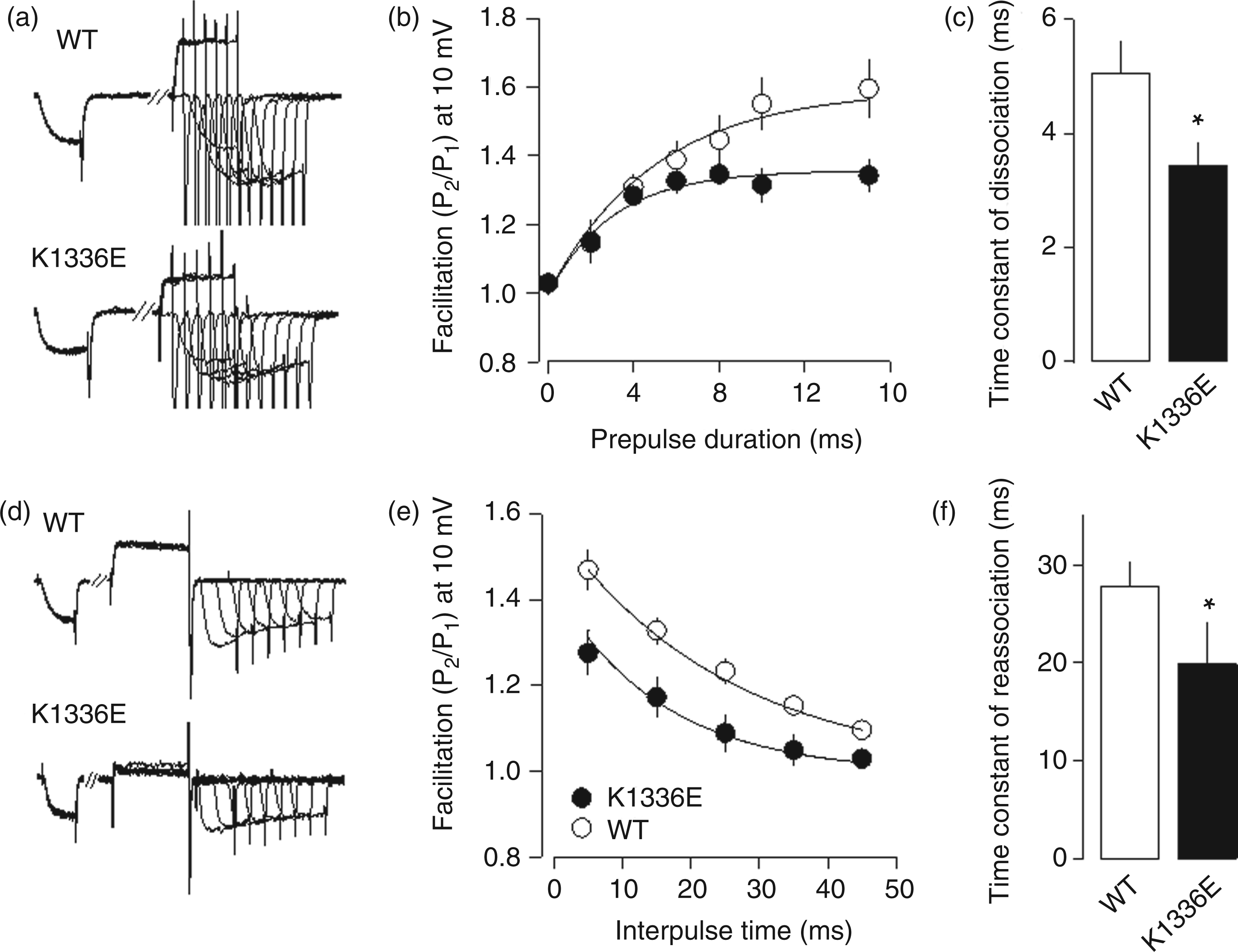
Although the reason for these differences in kinetics is presently unknown, it might be related to electrostatic potential changes occurring in the channels because of the presence of the mutation. Given that hydrophobic interactions and electrostatic complementarity are important for high-affinity interactions, we modeled domain III in the CaV2.1α1 subunit (where the K1336E mutation is located) and determined its electrostatic potential. Our analysis shows that the electrostatic potential was drastically affected by the K1336E mutation (Figure 4) because of the substitution of a positively charged amino acid (lysine) for a negatively charged one (glutamate).
Electrostatic potential distribution in domain III of the CaV2.1α1 channel subunit. (a) Ribbon diagram depicting S1–S6 transmembrane segments of domain III in the CaV2.1α1 subunit. The position of the K1336E mutation is denoted by the symbol. (b) The figure shows the most probable three-dimensional model of the CaV2.1α1 repeated domain III. The electrostatic potential of the analyzed region in the wild-type (WT) and in the mutant sequences (K1336E) is shown in red (positive) and blue (negative). The yellow arrows denote apparent changes in the electrostatic potential due to amino acid substitution. CaV2.1α1: neuronal voltage-gated P/Q-type Ca2+ channel.
Discussion
Previous work using heterologous expression systems has revealed changes in the biophysical properties of the P/Q-type channel induced by different FHM-1 mutations (11,12,19,26,27,33). In this work, we addressed the question of whether the K1336E mutation alters current density, and show that the density of functional channels in the cell membrane is reduced by the mutation. This alteration led to the prediction of decreased Ca2+ influx, and is consistent with previous findings suggesting a reduction of maximal current density through several FHM-1 mutant channels, including T666M, V714A, I1815L, W1684R and V1696I heterologously expressed in HEK-293 cells (11,12,27) as well as in neurons from the CaV2.1α1 null mouse (12,19).
However, in the present study we did not systematically address the changes induced by the K1336E mutant in current density or its effects on the targeting of the CaVα1-subunit to the plasma membrane, because such data would be difficult to interpret based on the results of previous studies in the R192Q knock-in mouse. Although heterologous expression has nicely revealed changes in mutant channel gating that can be confirmed in mutant mice in vivo (11,13,33), their role for determining effects on current density remains controversial (33).
Likewise, inhibition of P/Q-type channels by GPCRs is important for controlling neurotransmitter release. Though the complexity of this regulation is vast and involves distinct pathways that may converge on the channels, the characteristic features of this signaling mechanism include a reduction in current amplitude, a slowdown in activation kinetics, a shift in the voltage-dependence of activation and the development of prepulse facilitation during G-protein regulation. These characteristics have been incorporated into models in which the channels exhibit two functional gating states, “willing” and “reluctant” (34). The reluctant channels are bound to Gβγ and display the voltage-dependent shifts in channel gating noted above.
Previous studies have investigated the response to G-protein modulation of different FHM-1 mutant channels, and shown that the extent of G-protein-mediated inhibition and the prepulse facilitation is reduced by distinct FHM-1 mutations (25–27). Here, we studied the functional consequences of the K1336E mutation on direct G-protein regulation. This mutation is of particular interest not only because it is observed in some patients with hemiplegic migraine without cerebellar signs (2), but also because it resides in the extracellular face of the channel (Figure 1(a)), and the regions of the channel known to contribute to G-protein binding are located intracellularly. Our results show that activation of the hMOR receptor by DAMGO produced a substantial current inhibition through WT channels that was altered in the K1336E mutant channels. Our results also show that the mutation induced a reduction in prepulse facilitation. Among different explanations that could be tentatively proposed to explain these events, we have hypothesized that reversal of the charge at K1336E affects the electrostatic potential of both the extra- and intracellular surfaces of the cell membrane. Although this idea has yet to be experimentally tested (for instance by using additional CaV2.1α1 mutations with conserved charge changes as controls), the changes observed in G-protein-mediated regulation result in lowered affinity that occurs mainly through an increase in the channel/Gβγ dissociation rate koff as discussed below.
It is well established that the onset and decay of prepulse facilitation during Gβγ-induced inhibition can be represented by a simple two-state model (24,35,36)
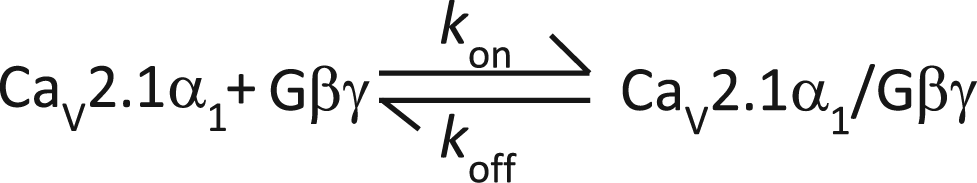
where CaV2.1α1 represents the closed state of the channel, kon is the association kinetic constant and koff is the dissociation kinetic constant. Our results revealed that the time constants for Gβγ dissociation were different for WT and mutant channels. The estimated off-rate (koff (s−1)) was ∼290 for the WT and ∼198 for the K1336E mutant channels. The estimated on-rate (kon (Gβγ) (s−1)) was ∼0.7 and ∼1 × 10−9 for the WT and K1336E channels.
On the other hand, in addition to mediating a voltage-dependent inhibition of CaV2.1 channels, a number of GPCRs trigger a concomitant inhibition of the channels via soluble second messenger pathways, which is called voltage-independent modulation (37). Interestingly, the increase in the fractional inhibition of the Ca2+ current with membrane depolarization suggests a possible contribution of voltage-independent modulation after DAMGO application. As shown previously, this modulation may have important functional implications. For example, activation of dopamine D1 receptors results in a protein kinase A (PKA)-dependent phosphorylation of protein phosphatase 1, which has been shown to dephosphorylate residues on P/Q-type channels (38). This illustrates that GPCRs mediate more than just voltage-dependent, membrane-delimited inhibition of CaV2 channels and raises the possibility of an altered cross-talk between voltage-dependent and voltage-independent pathways in the mutant channels. The contribution of changes in the voltage-independent modulation to the overall complexity of GPCR signaling to Ca2+ FHM-1 mutant channels is an interesting topic for future studies.
It is worth recalling that all data presented here were obtained in a heterologous system and that the functional implications of the K1336E mutation on native channels might be different. Unfortunately, the lack of a knock-in mouse (or other models) for this FHM-1 mutation hampers the study of these channels in its native environment. It should be noted, however, that the expression of cloned voltage-gated Ca2+ channels has proven to be a valuable research tool for studying the physiological properties of these proteins. In addition, the HEK-293 cells lack endogenous Ca2+ currents and provide a robust and reliable platform in which to express recombinant Ca2+ channels with high fidelity (39). Therefore, this cell line has been extensively used to examine the G-protein-mediated regulation of both wild-type and mutant Ca2+ channels. Indeed, an important part in our understanding of the molecular basis underlying migraine in general, and FHM-1 in particular, comes from studies using heterologous systems. However, the results obtained in the heterologous system need to be validated in a native system, and it will be interesting to see whether the mutant phenotype of CaV2.1 channel currents vary in cultured neurons isolated from different brain regions of K1336E transgenic mice when such animals are available.
Although at this moment it is still unclear to what extent the alterations provoked by the K1336E and other FHM-1 mutations play a role in the pathogenesis of the disease, a favored hypothesis considers neuronal hyperexcitability in the cerebral cortex as the basis for susceptibility to migraine. This would favor the onset of CSD, an abnormal increase of cortical activity that is followed by a long-lasting neuronal suppression wave that propagates across the cortex and is believed to initiate episodes of migraine with aura (4,7,23,40–42). It has been proposed that by an activation of the trigeminovascular system that results in alterations in the meningeal blood vessels and brainstem nuclei, CSDs lead to the development of headache. It should be mentioned, however, that there is evidence showing that vasodilation of meningeal and/or extracranial arteries may not be necessary or sufficient to cause migraine pain. Meningeal inflammation and other processes such as central sensitization or hypersensitivity to calcitonin gene-related peptide (CGRP)-mediated modulation of nociceptive pathways are now considered to be important mechanisms that may activate and sensitize perivascular meningeal afferents and lead to migraine pain (10). Last, although the effects of the K1336E mutations on overall neuronal excitability are difficult to predict, an increased postsynaptic excitability might result from this loss of function variants of presynaptic channels controlling release of excitatory neurotransmitters.
In conclusion, several mechanisms seem to contribute to the differential actions of FHM-1 mutations on CaV channel activity, including differences in G-protein modulation. Our data provide evidence suggesting that the K1336E mutation causes conformational changes in the CaV2.1α1 subunit that result in alterations in different biophysical properties of P/Q channels as well as in the Gβγ-mediated inhibitory pathway. Although these results are consistent with a causative role of the mutations in the disease, further mechanistic insights into the pathophysiology of FHM-1 are needed to support such a hypothesis.
Clinical implications
Familial hemiplegic migraine type 1 (FHM-1) is characterized by migraine, hemiparesis and ataxia. FHM-1 is linked to mutations in the gene coding the neuronal P/Q-type calcium channel. FHM-1 K1336E mutation affects the kinetics of dissociation and reassociation of the G protein from the channels. These studies shed new light onto the pathophysiology underlying FHM-1.
Footnotes
Acknowledgements
We appreciate the technical expertise of M. Urban and G. Aguilar. We thank Dr M. de Waard (Grenoble Institute of Neurosciences) and A. van den Maagdenberg (Leiden University) for generously providing us with the CaV2.1α1 constructs we used. We also thank the two anonymous reviewers for their insightful comments, which helped to make this a better manuscript.
Funding
This work was supported by grants from Conacyt to R.F. (128707) and PAPIIT to A.S. (IN221011). Fellowships from Conacyt to E.G.L., M.A.G and R.G.R. are gratefully acknowledged.
Conflict of interest
None declared.