
Abstract
Background
Short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing (SUNCT) and short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms (SUNA) are rare types of trigeminal autonomic cephalalgias (TACs).
Objective
To describe a series of patients with SUNCT and SUNA including relationship to pituitary tumors.
Method
All patients diagnosed with SUNCT or SUNA in the Calgary Headache Assessment and Management Program were reviewed.
Results
Six patients (five SUNCTs and one SUNA) were identified. The pain was severe, sharp, showed fixed-laterality, involved mainly the orbito-fronto-temporal region and was associated with autonomic symptoms. Attack duration ranged from 3 to 300 seconds and frequency was 1–200 paroxysms/day. MRI showed ipsilateral pituitary adenomas to the pain in five out of five of the SUNCT patients. Patients with adenomas underwent surgery. Pathology included three prolactinomas, and one mixed adenoma and gangliocytoma. One patient has remained headache free for 4 years after surgery. One was pain free for a year, and then headaches returned with tumor recurrence. Another had major improvement, and two have not improved. Patients were generally refractory to medications.
Conclusion
All five of our patients with typical SUNCT had pituitary tumors, with headache ipsilateral to the pituitary tumors in all cases. Tumor removal provided major improvement in three out of five patients. Medical treatment was only partially effective.
Introduction
Short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing (SUNCT) and short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms (SUNA) were first described in 1989 by Sjaastad et al. (1). SUNCT is classified under the trigeminal autonomic cephalalgias (TACs) in the Second Edition of the International Classification of Headache Disorder (ICHD II), and SUNA is described in the appendix (2). The diagnostic criteria of SUNCT requires at least 20 attacks of unilateral orbital, supraorbital or temporal stabbing or pulsating pain lasting 5–240 seconds. The pain must be accompanied by ipsilateral conjunctival injection and lacrimation and attacks occur with a frequency of three to 200 attacks per day (2). The diagnostic criteria of SUNA requires only one autonomic symptom and allows for a wider range of attack duration (from 2 seconds to 10 minutes).
From an Australian case series, the combined annual incidence of SUNCT and SUNA was estimated to be 1.2/100,000 (3). The prevalence of SUNCT/SUNA was estimated to be 6.6/100,000. Secondary SUNCT syndrome associated with pituitary micro- or macroadenoma has been reported (4–13). Patients with SUNCT represented approximately 5% of patients with headache and pituitary tumors (14). The pituitary tumors most commonly associated with headache were prolactinomas (37%) and growth hormone secreting tumors (33%) (14). A large series of patients with SUNCT from the UK reported that 8% of patients with SUNCT were found to have pituitary tumors on magnetic resonance imaging (MRI) (15).
We report here our case series of six patients with SUNCT and SUNA and discuss clinical phenotypes, neuroimaging findings, response to medical therapy, and the relationship of SUNCT and SUNA to pituitary tumors. These six patients represent our entire experience to date with these disorders, and remarkably, with careful neuroimaging, five of our six patients were found to have pituitary adenomas.
Methods
All patients with SUNCT/SUNA clinical features known to us in the Calgary Headache Assessment and Management Program in Calgary and the Calgary Chronic Pain Center were reviewed from 2003 to 2010. These programs provide a regional resource for Southern Alberta (a population of approximately 2 million) for difficult headache problems. All patients described here, except one, had been seen in consultation by the authors, and the attending neurologist of the other has been extensively communicated with. In addition, all charts have been carefully reviewed, as has the associated diagnostic imaging.
Results
Clinical characteristic of SUNCT and SUNA.
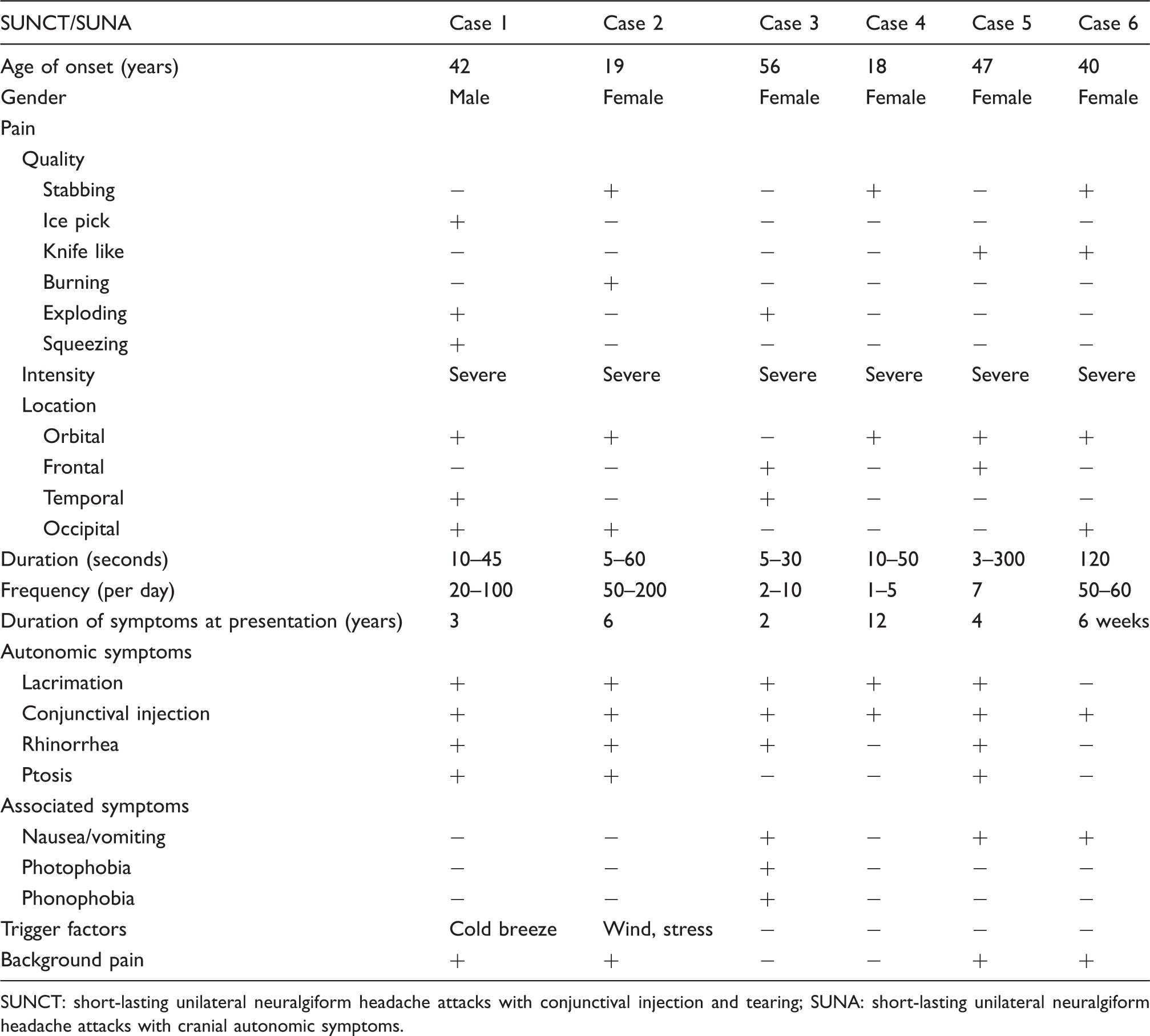
SUNCT: short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing; SUNA: short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms.
Relationship to pituitary tumor.

SUNCT: short-lasting unilateral neuralgiform headache attacks with conjunctival injection and tearing; SUNA: short-lasting unilateral neuralgiform headache attacks with cranial autonomic symptoms; MRI: magnetic resonance imaging; N/A: not applicable; IGF-1: insulin-like growth factor-1; RR: reference range.
Case reports
Case 1
A male patient presented at age 45 years, with a 3-year history of headache. He described an ice pick-like pain, which was exclusively right-sided, severe, and limited to the right temporal region. Pain paroxysms occurred over 20 times per day with a maximum of 100 per day, and would last up to 10–30 seconds. These would occur every day for several weeks at a time, and were associated with redness and tearing in the right eye. Initially, he had headache free periods every month lasting from a few days to a few weeks; however, gradually his headaches worsened and became daily. In addition, he described daily intermittent peculiar tight squeezing sensations which would begin occipitally on the right and move forward to cause a very tight painful squeezing or exploding sensation in the right retro-orbital region. These were unbearable, would last up to 45 seconds, and would occur 20 times a day, at times more often. They were also associated with redness and tearing of the right eye and even with ptosis when they were particularly severe. This headache could be brought on by inhaling cold air when he was outside in the winter. He also described a low grade headache with steady non-throbbing pain without associated nausea which was present all the time and involved primarily the occipital region bilaterally, occasionally the whole head. Interestingly, all headaches were improved with vigorous exercise such as playing hockey. MRI and computer tomography (CT) scans showed a large arachnoid cyst posterior to the cerebellum (3.4 × 8.7 cm) with some hypoplasia of the cerebellum.
He was prescribed many medications including lamotrigine (he could tolerate only very low doses), indomethacin (25 mg TID, concentration problems), gabapentin (300 mg per day, overly sedated), topiramate (50 mg per day, overly sedated), prednisone (70 mg per day), carbamazepine (400 mg per day), verapamil and a greater occipital nerve block. He was intolerant to essentially all medications and none provided significant improvement at the doses taken except for carbamazepine which provided minor relief for several months, but caused intolerable cognitive side effects. Subsequent MRI with sellar views showed a focal mass in the right side of the sella (1 × 1.1 × 1.5 cm), which was also present in retrospect on two previous MRI studies. There was no cavernous sinus invasion and the posterior fossa arachnoid cyst remained unchanged. There had been no interval change in size over 2 years, although previous MRI scans did not have specific pituitary views. A cine-magnetic resonance imaging (CINE MRI), cerebrospinal fluid (CSF) flow study showed no evidence for obstructed CSF flow at the foramen magnum. MRI of the cervical spine showed multilevel degenerative changes. Prolactin level was normal at 4 µg/l (normal range 0–25). Testosterone was slightly low at 7.7 nmol/l (normal range 8–29). Human growth hormone was normal at 1 µg/l (normal range 0–5), but insulin-like growth factor (IGF-1) levels were mildly elevated on two occasions at 489 and 373 µg/l (normal range 110–360). He had no clinical features of acromegaly. He underwent transsphenoidal removal of the pituitary adenoma in December 2006, 18 months after his presentation to us. The surgery relieved his headaches immediately, and there has been no recurrence of his SUNCT headaches over 4 years of follow-up. After surgery, he discontinued all medications for headache, including carbamazepine. Tumor pathology showed a mixed gangliocytoma and pituitary adenoma. The adenoma component consisted of sheets of chromophobe cells with only a very small minority staining positive for prolactin and adrenocorticotropic hormone (ACTH). A follow-up MRI in 2008 showed no tumor recurrence, and IGF-1 levels normalized at 203 µg/l after surgery.
Case 2
This female patient presented at age 25 with a 6-year history of headache. Initially, her headaches occurred once per week or less and consisted of a left-sided burning and stabbing pain which began occipitally and radiated to the retro-orbital region. The episodes of severe stabbing pain lasted between 5 and 60 seconds. Headache frequency increased gradually over several years, and became daily, with an attack frequency of approximately 50–200 per day. The pain became more limited to the area of the eye, and although between attacks she had some constant discomfort in her left eye and neck, it was not intense. Her attacks were associated with tearing, injection of the left eye and rhinorrhea of the left nostril. The left eye also showed swelling and ptosis at times. There was no associated nausea, photophobia, or phonophobia. Emotional stress and wind on her face could increase the frequency of her attacks. Physical examination showed slight ptosis of the left eyelid and tearing and injection of the left eye during the attacks. Her initial enhanced brain MRI with sellar views was interpreted as normal.
A trial of indomethacin (75 mg TID orally) provided no relief, and a diagnosis of SUNCT was made. Lamotrigine (400 mg per day), topiramate (200 mg per day), carbamazepine (600 mg per day), gabapentin (2400 mg per day), oxycodone and greater occipital nerve block were all ineffective. Intravenous lidocaine could not be arranged because of family issues. A brain MRI 2 years after the first one showed a 4-mm left pituitary mass without extrasellar extension. In retrospect, on review a previous brain MRI with sellar views showed an adenoma in the left lateral aspect of the pituitary gland measuring 2.5 × 3 mm. Her prolactin level was elevated at 40 µg/l. She underwent transsphenoidal pituitary tumor removal, and pathology showed an adenoma. The tumor cells stained positive for prolactin, and a small minority were positive for growth hormone. Postoperatively, the patient’s symptoms did not improve in over a year of follow-up.
Case 3
This female patient first experienced severe head pain upon awakening at age 56 years. After that, she experienced daily pain paroxysms limited to the right fronto-temporal region with an intensity of 7–9/10. She described these as ‘an explosion in her brain’. These episodes had duration of 5 to 30 seconds, occurred two to ten times per day, and were accompanied by ipsilateral rhinorrhea, tearing, redness of the eye, forehead sweating, and at times nausea/vomiting, photophobia, and phonophobia. She was completely pain free between pain paroxysms. The physical and neurological examinations were unremarkable. A brain CT arranged a few weeks after the onset of headache and subsequent brain MRI showed a subacute moderate-sized right parietal infarct. As a result, she was considered to have a silent brain infarction, and it was unclear whether the headache and stroke were related.
Medication trials with amitriptyline and indomethacin (75 mg TID) were not helpful. Carbamazepine was not tolerated. Subsequently lamotrigine (225 mg BID), prednisone (60 mg per day), verapamil (240 mg QID), prochlorperazine, meperidine, morphine, medical marijuana and botulinum toxin were tried without success. Pregabalin was attempted twice at 600 mg/day. It provided slight benefit by reducing pain intensity but this effect lasted only about a month on each occasion. Intravenous lidocaine in a dose of up to 4 mg/hour for 72 hours showed only a partial response and caused side effects. Repeat brain MRI with sellar views 6 years after headache onset showed a 4 × 6 mm right pituitary adenoma. In retrospect, it was present on the MRI study done previously. Her prolactin level was normal at 14 µg/l. She underwent transsphenoidal tumor removal but her headaches did not improve over 6 months of follow-up. Pathological examination did not reveal any diagnostic tumor tissue; likely due to suction aspiration of the tumor during surgery. Brain MRI 4 months post-surgery showed no residual tumor.
Case 4
This 30-year-old female patient presented to us in 2003 with a 12-year history of very severe pain above her right eyebrow. She also had a 7-year history of galactorrhea and a 5-year history of irregular menstruation. Initially her pain episodes were very infrequent, and occurred only three or four times a year. They gradually became more frequent until they were occurring a few times a week. The painful episodes lasted from 10 to 50 seconds and consisted of a single, brief, clearly localized pain, which was felt behind the right orbital region. They were associated with tearing and redness of the right eye. She was found to have an elevated prolactin level and a head CT showed a pituitary microadenoma. Brain MRI with sellar views subsequently showed a right 6-mm pituitary adenoma. Her hyperprolactinemia was treated with bromocriptine and cabergoline, neither of which the patient could tolerate. Prolactin level remained mildly elevated at 47 µg/l. She eventually underwent transsphenoidal tumor removal. At surgery, the pituitary adenoma could not be removed completely due to bleeding and she required blood transfusions. Following surgery, the pain disappeared for a year. It then recurred and became progressively more frequent until the attacks occurred on at least 5 days a week. The patient was referred to us due to recurrent headaches 4 years after surgery. She usually had one or two attacks per day with a maximum frequency of five per day. An enhanced brain MRI 4 years after surgery showed a recurrent 7-mm pituitary tumor on the right side.
Over the 20-year period during which she experienced these short-lived headache attacks, she was never free of them for any length of time except for 1 year after her pituitary surgery in 1999, and during her first pregnancy in 2005. Her second pregnancy did not ameliorate her headaches. Follow-up MRI 11 years after surgery showed further enlargement of her pituitary adenoma at 9 × 6 × 5 mm. Prolactin levels remained elevated at 49 µg/l 12 years after surgery. Because of the short duration of her headache attacks (10–50 seconds), she did not require medication for them and she refused to have a second operation.
In addition to the brief pain episodes described above, the patient also experienced another headache type, diagnosed as migraine, which was severe, bifrontal, and which occurred about twice a month, often before menstruation. There was associated nausea, and these attacks responded well to ibuprofen.
Case 5
A 51-year-old female patient presented with irregular menstruation, decrease in libido, galactorrhea and a 4-year history of episodic headache. The headaches were described as knife-like, 9–10/10 in intensity, involved the right frontal area just above the eyebrow, and lasted from a few seconds to 2 minutes. The attacks were infrequent at first, but they increased in frequency over time to several attacks an hour, averaging seven times per day. These exacerbations were accompanied by lacrimation and conjunctival injection of the right eye, and occasionally by right-sided ptosis and rhinorrhea. She had mild nausea and occasional vomiting, but no associated photophobia or phonophobia. Between pain paroxysms, she had a continuous fronto-occipital background headache, which involved the top half of her head with an intensity of 3/10.
Because the patient experienced episodes of right arm and facial numbness with her pain paroxysms, she was investigated with brain MRI, MRI angiogram, echocardiogram, and holter monitoring. No evidence of stroke or embolic source was found, but brain MRI showed an 11-mm right pituitary adenoma with a hemorrhagic component. Her serum prolactin level was elevated at 104 µg/l. Thyroid stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), IGF-1 and 24-hour urine cortisol levels were normal. Cabergoline 0.5 mg twice weekly resulted in a gradual reduction in her prolactin level and a slight decrease in the size of her pituitary adenoma on subsequent MRI, and reduced her headaches to some degree. Indomethacin (75 mg TID), gabapentin (1800 mg per day), and opiate analgesics were not helpful. Lamotrigine was discontinued because of a rash. Carbamazepine (800 mg per day) only gave slight benefit.
She underwent transsphenoidal resection of her pituitary adenoma in May of 2010, 5 years after headache onset. Cabergoline was discontinued a week prior to surgery. After surgery, her galactorrhea stopped and her headaches were greatly reduced in both intensity and frequency but did not disappear completely. She came off all pain medication, and her prolactin level normalized. Enhanced brain MRI 3 months after surgery showed scar tissue in the pituitary fossa but no evidence of adenoma. Her prolactin was normalized at 9 µg/l. Tissue pathology was reported as atypical prolactinoma, with the great majority of tumor cells staining positive for prolactin. After 18 months of follow-up, she continues to have major improvement in her SUNCT-like attacks, and experiences approximately one attack a week. These remaining attacks last from several seconds to 5 minutes. She is troubled now by some degree of constant right-sided headache which appears indomethacin-responsive, but indomethacin could not be continued because of concerns over renal dysfunction.
Case 6
A 40-year-old woman presented in 2008 with a 6-week history of severe, left-sided retro-orbital, knife-like headache. These headache attacks lasted approximately 2 minutes, and occurred every 10–15 minutes (approximately 50–60 per day). They were accompanied by congestion and redness of her left eye but no actual tearing. She had nausea with her headaches and at times would vomit. Her brain MRI demonstrated slight fullness of the pituitary gland but no evidence of adenoma. Initial differential diagnosis included paroxysmal hemicrania and SUNA syndrome. Indomethacin was titrated up to 75 mg three times a day but was not effective. A diagnosis of SUNA was made, and she was treated with lamotigine and carbamazepine, which gave no benefit and caused intolerable side effects. Diclofenac, ibruprofen, cyclobenzaprine, and tramadol with acetaminophen were also not helpful. Morphine provided only mild relief. Topiramate was not used because of a history of renal calculi.
Four months later, the patient was still having daily headaches. Therefore, verapamil was initiated and titrated up to 640 mg per day. This resulted in major improvement. She had only two orbital stabs a month even though the dose had to be reduced to 480 mg daily because of a prolonged PR interval. This improvement persisted for over 2 years of follow-up, but she developed a low-grade constant left-sided headache with some associated tearing and congestion of the left eye. This constant headache also did not respond to another trial of indomethacin. She was tender to palpation in an area in the left suboccipital region, where palpation caused pain to radiate to the left eye. The addition of left greater occipital nerve blockade (1% lidocaine plus 40 mg methylprednisolone) to the verapamil would render her completely headache free for up to 4 months. Three years after headache onset, she remains virtually pain free with this treatment.
Discussion
SUNCT and pituitary adenomas
SUNCT and SUNA are classified as primary headache types in the International Classification of Headache Disorders (ICHD) (2), but the SUNCT phenotype has been known to occur in patients with pituitary tumors (4–13) and other CNS lesions (16–23). Although the relationship of the headache syndrome to the pituitary tumor is not always clear, removal of the tumor has resulted in resolution of the SUNCT-like headaches in some cases (9–13). The ICHD acknowledges that the most common mimics of SUNCT are lesions in the posterior fossa or involving the pituitary gland (2).
A remarkable finding in our small case series is that all five patients with SUNCT syndrome were found to have pituitary adenomas, and the tumors were always ipsilateral to headache. Pituitary surgery was performed in an attempt to relieve headache in all cases except Case 4 who had surgery because of prolactinoma associated galactorrhea and menstrual abnormality before she was diagnosed with SUNCT syndrome. Three patients improved dramatically after adenoma removal. The pituitary adenomas were small, although two met criteria for macroadenoma (Cases 1 and 5). SUNCT improvement was greatest in patients with macroadenomas. The patients who did not respond to surgery tended to have smaller pituitary tumors (one non-secreting and the other with a prolactinoma with only mildly elevated prolactin levels).
Previous case series have shown a much lower prevalence of pituitary tumors in patients with the SUNCT phenotype. In one case series from Australia which included 17 patients with SUNCT and seven with SUNA, no pituitary lesions were found (3). In another large series from the UK with 43 SUNCT patients and nine SUNA patients, 8% of SUNCT patients were found to have pituitary lesions. No pituitary lesions were found in the patients with SUNA (15). The widely differing proportions of patients with SUNCT syndrome who are found to have pituitary adenomas may relate to at least two factors. One is patient selection, as in some regions patients found to have pituitary macroadenomas and SUNCT-like symptoms might not be referred to headache specialists. Another might be the extent to which patients with SUNCT are investigated. Pituitary adenomas are easily missed on neuroimaging. Three of our five patients had initial MRI scans reported as normal by experienced neuroradiologists, and the pituitary tumor was only discovered on subsequent scans, although it was present in retrospect on the initial scans in all of them. It is important that dedicated sellar views be performed in patients presenting with SUNCT if smaller pituitary adenomas are to be detected.
Pituitary tumors have been associated with a wide variety of headache types, including SUNCT (14). In a patient series of 84 patients with pituitary tumors and troublesome headache, 5% were found to have SUNCT, and all of these had either prolactinomas or growth hormone secreting tumors (14). This is consistent with our case series in that in our five cases of SUNCT associated with pituitary adenomas, two patients had pathologically proven prolactinomas, one had an atypical prolactinoma and a serum prolactin of over four times the upper limit of normal, and one patient had elevated serum IGF-1 levels, indication a growth hormone secreting tumor. Our third patient had a small adenoma and normal serum prolactin levels. Tumor pathology was not available on this patient, and she did not improve after surgery. This raises the possibility that in her case the relationship between SUNCT and her pituitary adenoma may have been purely a coincidental one. As well, this patient had a parietal lobe infarction closely related to the time of headache onset. It is possible that her stroke may have played a role in her headache rather than the pituitary adenoma (22,24). In a study involving high-resolution MRI scanning in 100 normal volunteers, 10% were found to harbor pituitary adenomas (25). A systematic review on the prevalence of pituitary adenomas, considering both autopsy and radiologic studies, estimated that the prevalence of pituitary adenomas was even higher, at 16.7% (26). These data would suggest that even if some types of pituitary adenomas have a direct causative role in producing SUNCT-like symptoms, there would still remain a proportion of patients with primary SUNCT where an incidental pituitary adenoma is found. In fact, if it were assumed that true (primary) SUNCT had a pathophysiology with no relationship to pituitary adenoma, one would still expect to find incidental pituitary adenomas in at least 10% of patients. Such patients would not be expected to improve with therapies including surgical therapies directed specifically at the adenoma, and our Case 3 may well be an example of such a patient.
The great majority of published case reports and case series in the medical literature that report an association between pituitary and SUNCT indicate that the adenoma involved was either a prolactinoma or a growth hormone secreting tumor (4–14). Another case series with two additional patients included a microprolactinoma. The pathology of a patient with a macroadenoma was not specified (15). It would appear, then, that virtually all pituitary adenomas associated with SUNCT including all but one of the patients in our series are associated with prolactin or growth hormone secreting tumors. Endocrinological testing including serum prolactin and IGF-1 levels can therefore be useful as the investigation of patients with SUNCT, and the MRI can be expedited if these results are abnormal. Rocha Filho et al. (11), however, reported a patient with a non-secreting pituitary adenoma with SUNCT. Although one might consider the relationship in their patient to possibly be coincidental, unlike our one similar case (Case 3), the patient in their case report remitted after surgery and remained symptom free during 14 months of follow-up.
Symptomatic SUNCT and headache mechanisms
Given the wide range of possible causes of symptomatic or secondary SUNCT, it would seem clear that several different mechanisms must be able to produce this clinical syndrome. It is unclear how a pituitary adenoma, particularly a microadenoma, could result in SUNCT symptoms, but it may be relevant that, as in our series, the headache is almost always ipsilateral to the tumor. With regard to pituitary tumors and headache in general, although localized dural stretch and cavernous sinus invasion have been postulated as possible mechanisms (27,28), these are not satisfactory mechanisms for most patients with pituitary microadenomas. Intrasellar pressure was identified as a possible mechanism for headache production by Arafah et al. (29). Intrasellar pressure was measured at the time of transsphenoidal surgery in 49 patients with pituitary adenoma, and many showed elevated intrasellar pressures. Patients presenting with headache had higher mean intrasellar pressures than those who did not, regardless of tumor size. Interestingly, in another study of 63 patients with pituitary tumors, 70% of whom had headache, there was no positive correlation between clinical headache score and pituitary volume (30). Two studies involving 114 patients in total could find no correlation between cavernous sinus invasion and headache (30,31), and the second study (31) also could find no correlation between tumor size and headache. Patients with SUNCT, however, make up only a small proportion of patients with pituitary tumors and headache, and the relevance of these findings to the pathophysiology of SUNCT secondary to pituitary tumors is unclear. The unusual headache pain of SUNCT may have mechanisms quite different from other headache types.
It might be expected that derangements in hormonal function and hypothalamic neurotransmitters would play a role in SUNCT associated with pituitary adenoma. Dopamine may play role in the pathophysiology of primary headache disorders (32) and dopamine is also an important hypothalamic neuroendocrine inhibitor, playing a role in the regulation of prolactin secretion from the anterior pituitary gland (33). Many patients with SUNCT attacks and pituitary tumors have prolactinomas, and prolactin may therefore play a role in the pathogenesis of SUNCT attacks. One unusual patient with a pituitary microadenoma and SUNCT attacks which occurred only nocturnally was shown to have nocturnal elevations of prolactin levels which coincided with the headache symptoms (34). Patients with SUNCT and associated prolactinomas have been reported who suffered major exacerbations of headache symptoms after dopamine agonist administration (4,9,12). Indeed, patients with prolactinomas without headache have suffered SUNCT symptoms for the first time after cabergoline (6,7). On the other hand, a patient with prolactinoma and SUNCT has been reported in whom administration of dopamine agonists alleviated headache symptoms (5).
Clomiphene citrate, presumably through its effect on the hypothalamus, has also been reported to be effective in the treatment of SUNCT (35). The resolution of SUNCT headache symptoms after removal of growth hormone secreting pituitary microadenomas supports a potential role for growth hormone in SUNCT pathogenesis (10,13).
Functional magnetic resonance imaging (fMRI) in spontaneous SUNCT attacks has demonstrated activation of the ipsilateral hypothalamic gray (36). A recent study utilizing blood oxygen level dependent (BOLD) changes in fMRI showed variability among patients with primary SUNCT, with some showing bilateral activation of the hypothalamus, some only contralateral activation, and some showing ipsilateral negative activation. SUNA patients showed negative activation bilaterally, whereas there was no activation in symptomatic SUNCT (37). Clearly more data are needed before the role of the hypothalamus in SUNCT can be understood.
Medical treatment
The EFNS task force has proposed that the medical treatment of choice for SUNCT and SUNA is lamotrigine, followed by topiramate and gabapentin (38), although medical treatment is often disappointing in clinical practice. Based on case reports, lamotrigine appears to be the most effective medication for SUNCT (8,38–43). Other treatment options include gabapentin, topiramate, verapamil, carbamazepine, oxcarbazepine, zonisamide, intravenous lidocaine, intravenous phenytoin, and greater occipital nerve block (19,44–59). Intravenous lidocaine has been reported to completely abolish the pain in many patients with both SUNCT and SUNA. The effect of intravenous lidocaine can last up to 6 months after infusion (37). As a prophylactic medication, lamotrigine has been reported to show a response rate in SUNCT of 68–100%, and topiramate a response of 52% (3,37). However, patients with episodic SUNCT or SUNA are generally more responsive to lamotrigine than the chronic forms. Gabapentin has shown benefit in patients with SUNA (15). Greater occipital nerve block has also been reported to show a beneficial effect in SUNCT but not in SUNA (37). In general, the efficacy of verapamil in SUNCT/SUNA syndrome is unclear. One case of SUNCT has been reported that responded well to verapamil (51). On the other hand, another case report found that verapamil used for hypertension precipitated SUNCT (60). Our patient with primary SUNA had a significant response to verapamil and has remained on this medication for several years. As the verapamil has not been stopped, we cannot be certain whether it was effective, or if the patient improved spontaneously. However, as the improvement has persisted for several years, we feel the verapamil was effective. In our patients with SUNCT and associated pituitary adenomas, two patients had a partial response to carbamazepine. Published case reports indicate that SUNCT can be responsive to both carbamazepine (53) and oxcarbazepine (54). One of our patients also showed some response to pregabalin, perhaps not surprising given that responsiveness to gabapentin has been reported (19,44–47). However, most reports on the use of prophylactic medications in SUNCT have discussed primarily patients with primary SUNCT. Patients with secondary SUNCT have generally been refractory to medical treatment.
Conclusion
A significant proportion of patients with the clinical symptoms of SUNCT have associated pituitary adenomas, particularly prolactinomas and growth hormone secreting tumors. There is evidence that this relationship is not coincidental, and tumor removal can result in dramatic improvement in the headache. In addition to the three patients in our case series who improved, six other cases of secondary SUNCT have been reported with significant headache resolution after surgical removal of pituitary adenomas (9–13). The high proportion of SUNCT patients with pituitary tumors found in our small case series may not be representative of the actual prevalence of pituitary tumors in patients with SUNCT. However, careful assessment for a pituitary adenoma is warranted in all patients with SUNCT, and based on our experience, unless very carefully looked for with dedicated MRI sellar views, these tumors are easily missed on initial neuroimaging.
Clinical implications
Many patients with SUNCT have pituitary adenomas. Headache is ipsilateral to the pituitary tumor. Pituitary adenoma is easily missed on MRI. Careful assessment is warranted. Adenoma removal provides significant improvement in some patients particularly those with functioning macroadenomas.
Footnotes
Acknowledgements
The authors thank Dr Jeptha Davenport and Dr Lara Cooke, Department of Clinical Neurosciences, Faculty of Medicine, University of Calgary, and Dr Alan Jackson, Faculty of Medicine, University of Manitoba, for their comments and discussion in the preparation of this manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.