
Abstract
Aim:
To quantify and compare the responsiveness within the meaning of clinical relevance of efficacy endpoints in a clinical trial with over the counter (OTC) analgesics for headache. Efficacy endpoints and observed differences in clinical trials need to be clinically meaningful and mirror the change in the clinical status of a patient. This must be demonstrated for the specific disease indication and the particular patient population based on the application of treatments with proven efficacy.
Methods:
Patient’s global efficacy assessment during two study phases (pre-phase and treatment phase) was used to classify patients as satisfied or non-satisfied with the efficacy of their medication. The analysis is based on 1734 patients included in the efficacy analysis of a randomized, placebo-controlled, double-blind, multi-centre parallel group trial with six treatment arms. Based on this classification and the pain intensity recorded by the patients on a 100 mm visual analogue scale, group differences by assessment categories and receiver operating characteristic (ROC) curve methods were used to quantify responsiveness of the efficacy endpoints ‘time to 50% pain relief’, ‘time until reduction of pain intensity to 10 mm’, ‘weighted sum of pain intensity difference’ (%SPIDweighted), ‘pain intensity difference (PID) relative to baseline at 2 hours’, and ‘pain-free at 2 hours’.
Results:
Clinically relevant differences between patients satisfied and non-satisfied with the treatment were observed for all efficacy endpoints. Patients with the highest rating of efficacy had the fastest and strongest pain relief. In comparison, patients assessing efficacy as ‘less good’ reached a 50% pain relief on average nearly an hour later than those scoring efficacy as at least ‘good’. Simultaneously, their extent of pain relief was only half as great 2 hours after medication intake. Patients scoring efficacy as ‘poor’ experienced practically no pain relief within the 4 hour observation interval. ROC curve calculations confirmed an adequate responsiveness for all continuous endpoints. The following cut-off points for differentiating between satisfied and non-satisfied patients were deduced from the data in the pre- and treatment phase, respectively: ‘time to 50% pain relief’ 1:10 and 1:31 h:min, ‘time until reduction of pain intensity to 10 mm’ 2:40 and 3:00 h:min, ‘%SPIDweighted’ 68 and 64%, ‘PID at 2 hours’ 35 and 35 mm. The sensitivity and specificity based on these cut-off points ranged from 70 to 79%. The binary endpoint ‘pain-free at 2 hours’ showed a clearly higher specificity (80 and 87%) than sensitivity (65 and 61%) in the pre- and treatment phase, respectively.
Conclusions:
When global assessment of efficacy by the patient was used as external criterion, ROC curve calculations confirmed a high responsiveness for all efficacy endpoints included in this study. Clinically relevant differences between patients satisfied and non-satisfied with the treatment were observed. The endpoint ‘%SPIDweighted’ proved slightly but consistently superior to the other endpoints. SPID and %SPIDweighted are not easy to interpret and the time course of pain reduction is of high importance for the patients in the treatment of acute pain, including headache. The endpoint ‘pain-free at 2 hours’ showed the expected high specificity, but at the cost of a concurrently low sensitivity and clearly makes less use of the available information than the endpoint ‘time to 50% pain reduction’, which combines the highly relevant aspects of time course and extent of pain reduction. Responsiveness, the ability of an outcome measure to detect clinically important changes in a specific condition of a patient, should be added in future revisions of IHS guidelines for clinical trials in headache disorders.
Keywords
Introduction
Headache pain is difficult to define and quantify. Consequently, quantification of analgesic efficacy is also difficult, in particular in clinical trials (1). The selection of primary efficacy endpoints in headache studies is a critical factor (2). In the 1970s and 1980s attention was focused on the benefits and drawbacks of the various methods for measuring pain, ranging from yes/no responses through graded scales with variable numbers of categories to the continuous visual analogue scale (3–6), but the debate has since come to centre on the question of adequate primary efficacy endpoints or outcome measures (2,7–10). During this period the discussion on headache study methodology concentrated on the methods used in the clinical development programme of sumatriptan, including the 4-stage Likert scale used as a method of measurement and the efficacy endpoint selected (‘percentage of patients with a reduction in headache severity from moderate or severe to none or mild’) (11–13). The Committee on Clinical Trials in Migraine of the International Headache Society suggested as the primary efficacy parameter in acute treatment trials in migraine the ‘number of migraine attacks resolved within 2 hours’ (14), although this suggestion has received some criticism in comparison with the response criterion referred to above (15). It has been noted out on several occasions, however, that little research has been conducted to determine which of these endpoints are considered by headache sufferers themselves to be most important (16,17), and it has also been commented that new efficacy endpoints need to be defined for certain patients (18).
The issue of which endpoint to use is still vexing (7). There is no consensus regarding how an endpoint translates into patient acceptability, or about the relative importance of each attribute in determining this acceptability (19). It is, however, universally accepted that in the context of clinical trials, outcome variables and observed differences need to be clinically meaningful (20) and mirror the change in the clinical status of a patient.
We used methods described for the assessment of outcome measures (21) to quantify and compare the performance of efficacy endpoints in clinical trials with OTC analgesics in headache. In general, outcome measures have to be reliable, valid and responsive. In a previous paper (22) we reported some results on the validity of various efficacy endpoints in OTC headache trials. In this paper we focus on the responsiveness of these efficacy endpoints.
Responsiveness is defined as the ability of an outcome measure to detect clinically important changes in a specific condition of a patient (21,23,24). An outcome measure with high responsiveness should be able to discriminate between the true condition states of the patient. If we restrict to two states of a condition (present or absent), this allows evaluation of responsiveness with methods originally used to assess the performance of diagnostic tests. The condition to be ‘diagnosed’ (25) could be, for example, whether the clinical status was improved or non-improved or whether the patient was satisfied or non-satisfied with their treatment. The quantification and comparison of responsiveness of various outcome measures necessitates the definition of an external criterion for the differentiation between patients with the condition present or not. There is currently no gold standard for an external criterion in pain outcome measures. Examples of external criteria used comprise pain assessment and disability rating (23), return to full activities (25), use of additional rescue medication (26), global perceived effect assessed by the patient (24), standardized effect size (27), power of a test or sample size needed to detect a clinically important difference (21) or patient’s global impression of change (28). We used the patient’s global assessment of efficacy as an external criterion to compare the responsiveness of common efficacy endpoints in headache trials.
Methods
Patients, study design and treatments
The data for this analysis were collected as part of a randomized, placebo-controlled, double-blind, multi-centre parallel group trial with six treatment arms, conducted between September 1998 and January 2003 (29). The primary objective of the study was to investigate the efficacy, safety and tolerability of the fixed combination of acetylsalicylic acid + paracetamol + caffeine in comparison with the combination without caffeine, the single preparations, and placebo in patients who were used to treating their episodic tension-type headache or migraine attacks with non-prescription analgesics.
The patients were enrolled by practitioners and specialists in general and internal medicine throughout Germany. Male or female patients (18–65 years) who were not consulting for headache were asked whether they had headaches that they treated with non-prescription analgesics. Usual headaches had to meet International Headache Society criteria (30) for episodic tension-type headache (2.1) and/or migraine with or without aura (1.1, 1.2.1). They must have experienced these headaches for at least 12 months with a minimum of two headache episodes within the previous 3 months.
Patients were excluded if previous or concomitant diseases or medication could interfere with one of the study drugs or influence headache symptoms. Drug overuse connected with the headache and alcohol or drug abuse was also an exclusion criterion, as was pregnancy, lactation or participation in another clinical trial within 4 weeks of entering this study.
Before enrolment the patients gave their written informed consent according to paragraphs 40 and 41 of the German Drug Law (AMG) and International Conference on Harmonisation, Guidance for Good Clinical Practice, E6 (ICH GCP) standards. Patients were allowed to terminate participation in the trial at any time, without giving reasons. The study was conducted in accordance with the Declaration of Helsinki, the AMG and ICH GCP standards and did not start before independent ethics committee approval was obtained.
Patients randomly allocated to one of the six treatment groups treated their headache attack with a single dose of the allocated study medication. Before the randomized treatment phase a headache episode treated with the patient’s usual non-prescription medication was recorded (open pre-phase).
Endpoints
Patients recorded pain intensity on a 100 mm visual analogue scale (VAS) before and then 30 min and 1, 2, 3 and 4 hours after drug intake in the patient diary. The calculated time to 50% pain relief was chosen as primary endpoint based on the pain intensity recorded on the VAS.
The secondary endpoints of this study, which comprise both efficacy and tolerability parameters, were:
calculated time until reduction of pain intensity to 10 mm on the VAS percentage of patients with at least 50% pain relief after 30 min, 1, 2, 3 and 4 hours (evaluated on the VAS) percentage of patients pain-free, defined as patients with reduction of pain intensity to at least 10 mm on the VAS, after 30 min, 1, 2, 3 and 4 hours pain intensity difference after 30 min, 1, 2, 3 and 4 hours (evaluated on the VAS) weighted sum of pain intensity difference (SPID) expressed as a percentage of the maximum achievable SPID (%SPIDweighted) extent of impairment of daily activities before and after 30 min, 1, 2, 3 and 4 hours of drug administration (4-point verbal rating scale (VRS): 0 = ‘not impaired’, 1 = ‘somewhat impaired’, 2 = ‘greatly impaired’, 3 = ‘usual daily activities impossible’) global assessment of efficacy by the patient (4-point VRS: 1 = ‘very good’, 2 = ‘good’, 3 = ‘less good’, 4 = ‘poor’) within 12 hours after administration of the trial medication based on the question ‘How do you assess the efficacy of your tablets?’ global assessment of tolerability by the patient and investigator (4-point VRS: 1 = ‘very good’, 2 = ‘good’, 3 = ‘less good’, 4 = ‘poor’) safety assessment: recording of adverse events (time of onset, duration and intensity of adverse events; relationship between the drug treatment and adverse event determined by the investigator)
Efficacy and safety endpoints were calculated twice, for the assessments at the end of the open pre-phase and for the assessments at the end of the randomized treatment phase.
Statistical analysis
The efficacy endpoints were compared by means of descriptive statistics sorted by the categories of the patient’s global assessment of efficacy.
The performance of the endpoints in discriminating between patients satisfied and patients non-satisfied with the efficacy was further evaluated using receiver operating characteristic (ROC) methodology (31). Patients assessing efficacy as very good or good were classified as satisfied, patients scoring efficacy less good or poor as non-satisfied. Subsequently and for each continuous endpoint separately, cut-off points on the measurement scale for the endpoint were deduced from ROC curves using logistic regression analysis. If the patient was classified as satisfied with the efficacy and the endpoint measurement was above the cut-off point, the outcome was called true positive (Figure 1A). If the patient was classified as non-satisfied and the endpoint measurement was below the cut-off point, the outcome was called true negative. The ROC curve displays the true positive (sensitivity) versus the false positive (one minus specificity) rates for the range of possible cut-off points for predicting the global assessment of efficacy by the patient (Figure 1B and C). The area under the ROC curve (AUC) was calculated as a summary measure of its discriminatory ability. The cut-off point was chosen where the sensitivity and specificity were equal assuming equal importance of sensitivity and specificity.
ROC method. (A) Decision table with possible outcomes. (B) Distribution of data of the efficacy endpoint for the two groups of patients with efficacy assessment as non-satisfied or satisfied including a possible cut-off point. (C) ROC curve for all possible cut-off points.
As the endpoint ‘pain-free’ at a pre-specified time point is a binary variable a ROC curve cannot be calculated. There is only one pair of sensitivity and specificity.
All calculations were performed twice based on the data of the pre-phase and the data of the randomized treatment phase.
Results
Patient characteristics
The full analysis set comprised 1743 patients recruited in 133 centres (29). Of these, 15 patients in the pre-phase and nine patients in the randomized treatment phase did not assess the global efficacy at the end of the respective study phase. The remaining 1728 patients in the pre-phase and 1734 patients in the randomized treatment phase were included in the evaluation (76% women, 24% men; median age: 38 years; range 16–72 years). Without treatment, the usual pain intensity was severe or very severe in 62% and moderate in 37% of patients. The severity of pain was associated with disability of performing usual daily activities. The mean ± SD pain intensity at baseline was 59.1 ± 20.6 mm in the open pre-phase and 64.3 ± 20.3 mm in the randomized treatment phase.
Major efficacy results
The superior efficacy of the triple combination containing acetylsalicylic acid, paracetamol and caffeine could be shown for all efficacy endpoints such as the ‘time to 50% pain relief’ (primary endpoint), ‘time until reduction of pain intensity to 10 mm’, ‘pain intensity difference’, ‘%SPIDweighted’, ‘extent of impairment of daily activities’, and ‘patient’s global efficacy assessment’ (29).
Group differences sorted by the assessment of efficacy
Summary of descriptive statistics for primary and secondary efficacy endpoints grouped by patient’s global efficacy assessment.
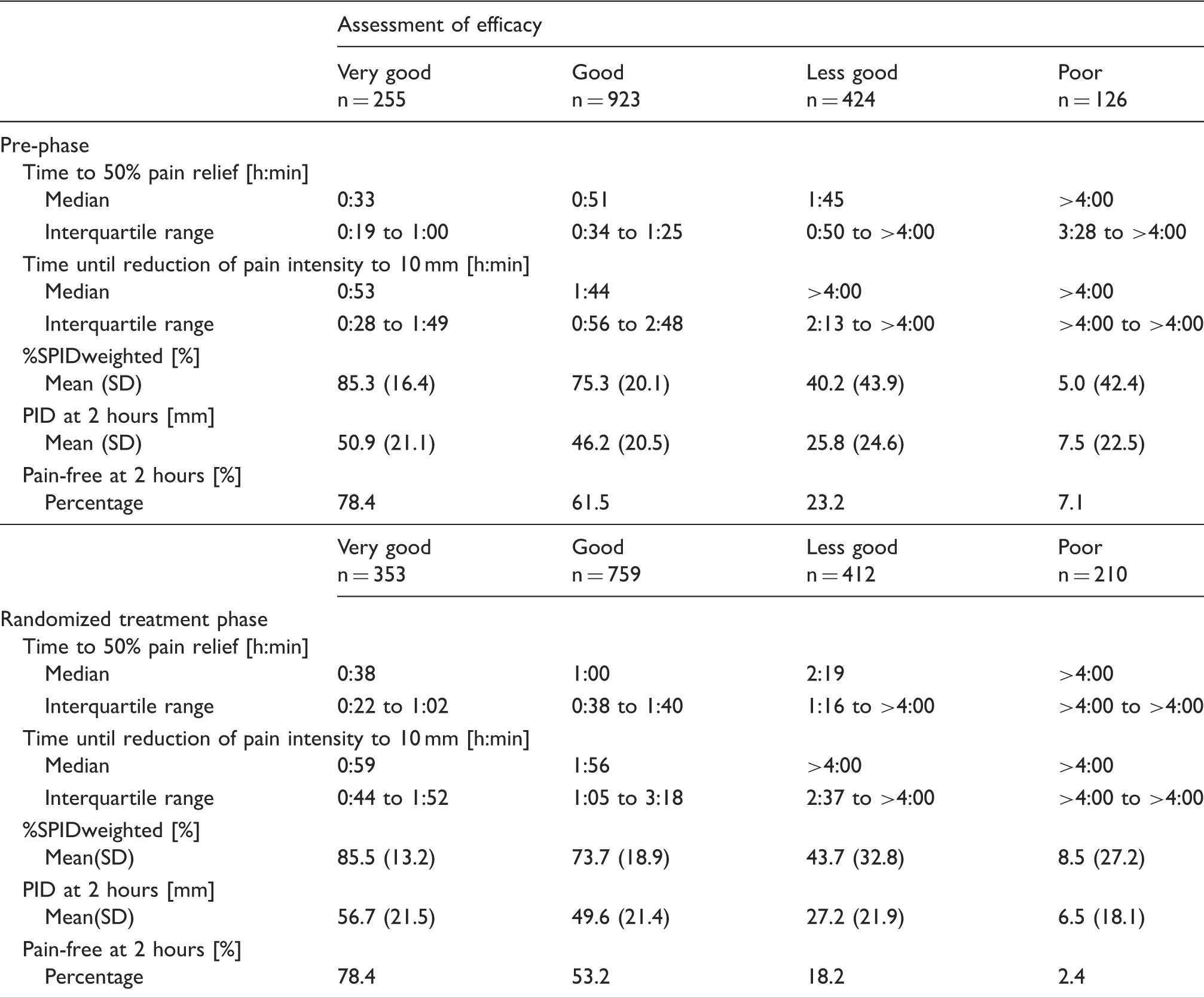
All efficacy endpoints improved in parallel to the increase of the patient’s efficacy assessment (Table 1). Patients with the highest rating of efficacy had the fastest and strongest pain relief. In comparison, patients assessing efficacy as less good reached a 50% pain relief on average nearly an hour later (median time to 50% pain relief 1:45 h:min) compared with those scoring efficacy as at least good (median time to 50% pain relief 0:51 h:min) in the pre-phase. Simultaneously, the extent of pain relief was only half as great 2 hours after medication intake (mean PID 25.8 mm versus 46.2 mm as assessed on the VAS). Patients scoring efficacy as poor experienced almost no pain relief within the 4 hour observation period (median time to 50% pain relief > 4 hours and mean PID 7.5 mm).
The corresponding values in the randomized treatment phase were overall slightly worse regarding the time to pain relief and slightly better with respect to the extent of pain relief (Table 1). The differences between the categories of the patient’s global assessment of efficacy were qualitatively and quantitatively well comparable to those in the pre-phase.
ROC curves and cut-off points
The ROC curves of all continuous efficacy endpoints were very close together and partly crossing (Figure 2). The ROC curves, the sensitivity and specificity, and consistently the AUC were more similar for the data from the randomized treatment phase than those from the pre-phase for all endpoints (Table 2). The AUC in the pre-phase ranged from 0.77 to 0.86 and that in the treatment phase from 0.84 to 0.89. The endpoint ‘%SPIDweighted’ was slightly but consistently superior to the other endpoints.
Receiver operating characteristic (ROC) curves for primary and secondary efficacy endpoints based on patient’s global efficacy assessment as external criterion. Area under the receiver operating characteristic (ROC) curve and sensitivity and specificity of cut-off points for primary and secondary efficacy endpoint based on patient’s global efficacy assessment as external criterion.

The optimal cut-off points for differentiating between satisfied and non-satisfied patients were lower in the pre-phase than in the randomized treatment phase for the majority of endpoints. The following cut-off points were deduced from the ROC curves for the pre-phase and treatment phase, respectively: ‘time to 50% pain relief’ 1:10 and1:31 h:min, ‘time until reduction of pain intensity to 10 mm’ 2:40 and 3:00 h:min, ‘%SPIDweighted’ 68 and 64%, ‘PID at 2 hours’ 35 and 35 mm. Based on these cut-off points the sensitivity and specificity ranged from 70 to 77% in the pre-phase and 76 to 79% in the treatment phase. The binary endpoint ‘pain-free at 2 hours’ showed a clearly higher specificity of correctly predicting a non-satisfied patient (80 and 87% for the pre-phase and treatment phase, respectively) than the sensitivity of correctly predicting a satisfied patient (65 and 61%).
Discussion
Responsiveness must be demonstrated for the specific disease indication and the particular patient population based on the application of treatments of proven efficacy (32). The data used was taken from a clinical study that showed the superior efficacy of the fixed combination containing acetylsalicylic acid, paracetamol and caffeine over the combination without caffeine, the single preparations, and placebo in the treatment of headache for all efficacy endpoints, such as the ‘time to 50% pain relief’ (primary endpoint), ‘time until reduction of pain intensity to 10 mm’, ‘pain intensity difference’, ‘%SPIDweighted’, ‘extent of impairment of daily activities’, and ‘patient’s global efficacy assessment’ (29).
The quantification of the responsiveness always necessitates the choice of a reference criterion that describes the status or change in a status of a patient’s condition. There is currently no gold standard for this criterion in pain outcome measures. The choice of the criterion is always problematic (24). It should be specific to the disease and the patient population studied. In self-medication, the patient’s choice of a particular therapy, which they take for their headache, depends on the subjective perception of efficacy and tolerability of the drug without consulting a doctor. As the global assessment of overall efficacy by the patient aims to summarize the patient’s overall impression about their state or change in their state (33), it is of particular importance in clinical trials with OTC medications for self-medication of headaches and qualifies as a criterion with which to quantify and compare the performance of efficacy endpoints in these trials. It is relevant and sensible to ask the patient to assess their perceived benefit (24) and to use their decision as a reference. The global assessment of efficacy in clinical trials reflects these perceptions best. However, as we do not postulate global assessment of efficacy as the ‘gold standard’, further comparisons of efficacy endpoints against other reference criteria may be helpful.
When global assessment of efficacy by the patient was used as external criterion, we have shown clinically relevant differences between patients satisfied and non-satisfied with the treatment for all efficacy endpoints included in the analysis. Patients satisfied with their medication reported a 50% pain relief approximately within 1–1.5 hours. A pain-free state should be reached by at least 3 hours after intake of the medication for patients to be satisfied with their medication. The reduction in pain intensity 2 hours after medication intake needed to be greater than 35 mm for these patients assessed on the 100 mm VAS.
The cut-offs for differentiating between patients satisfied and non-satisfied with their treatment were determined assuming equal sensitivity and specificity. This was possible for all continuous endpoints. The binary endpoint ‘pain-free at 2 hours’ used an a priori fixed cut-off point with clearly higher specificity of correctly classifying non-satisfied patients compared with the sensitivity of correctly classifying satisfied patients. Nearly balanced sensitivity and specificity would result in the binary outcome pain-free at 3 hours. Higher specificity than sensitivity could be reached for the other endpoints if more stringent cut-off points were chosen. Outweighing specificity or sensitivity might be meaningful for specific study objectives. Without any restrictions it is reasonable to balance both.
Lipton stated ‘The assessment of migraine pain, associated symptoms, and disability is subjective, in that clinicians rely on patient rating of the severity of migraine symptoms. Patient assessment and corresponding physician evaluation form the basis for treatment decisions and assessment of the efficacy of a migraine therapy’ (34). The FDA added, ‘For some treatment effects, the patient is the only source of data. For example, pain intensity and pain relief are the fundamental measures used in the development of analgesic products. Many patient-reported outcome instruments are able to detect mean changes that are very small; accordingly it is important to consider whether such changes are meaningful’ (35). Although there are some surveys that judge and rank the relevance of possible endpoints from the perspective of the patients (36–39), there is no clinical trial in headache to our knowledge in which the specificity and the sensitivity of the primary endpoint were analysed quantitatively with the global assessment of efficacy of the patients as reference criteria. This obvious question remained unanswered. Corresponding analyses would be helpful, but needs access to individual patient-related information.
With migraine and tension-type headache pain intensity increases over a certain period of time until fully developed. If the medication is taken very early, endpoints related to the baseline value are not very useful. This can be a problem in early intervention studies. The patients in the Thomapyrin study (29), however, were not instructed to take their study medication at any certain time point and the baseline values and the development of pain intensity show that in most patients the headache pain was fully developed.
The ROC curve calculations confirmed a high responsiveness for all efficacy endpoints included in this study. The observed differences between endpoints are, in general, small. Therefore the ROC curves for all endpoints were very close and partly crossing, although the endpoint ‘%SPIDweighted’ proved slightly but consistently superior to the other endpoints.
As recommended in the 2nd edition of the ‘Guidelines for controlled trials of drugs in migraine’ the ‘percentage of patients pain-free at 2 h, before any rescue medication, should usually be the primary measure of efficacy’ (40). According to the authors of the comments on this recommendation, this endpoint has the advantage that it ‘reflects patients’ expectations, is simple and not affected by rescue medication’ (40). The value of the 2-hour pain-free measure cannot be overemphasized, according to Ramadan (41). Although migraine patients stated incomplete or inconsistent pain relief as important issues for their assessment of a treatment in a telephone survey (36), the Thomapyrin study (29) showed that at least for analgesics used for the self-medication of headaches the patients weighted ‘time to 50% pain reduction’ much higher than ‘time to pain-free’ for their global evaluation of efficacy (22). Goadsby stated as a disadvantage that for patients with slowly settling headache the transition to no pain is difficult to discern and for some patients the reduction in headache pain to mild is a substantial and very beneficial result (7). Tfelt-Hansen and coauthors say in the guideline that ‘resolution, not alleviation, within 2 h might seem unrealistic with some drugs’ (40). The endpoint ‘pain-free at 2 hours’ showed the expected high specificity, but at the cost of a concurrently low sensitivity. It clearly makes less use of the available information than, for example, the endpoint ‘time to 50% pain reduction’ and copes less well with the objective ‘to choose appropriate endpoints that reflect realistic treatment goals for individual patients’ (42).
It was recommended in the 2nd edition of the ‘Guidelines for controlled trials of drugs in tension-type headache’ again that ‘pain-free rate at 2 h should be the primary efficacy measure’ (43). However, other possible endpoints were also discussed: ‘Sum of pain intensity differences’ (SPID) could theoretically be useful because it has the advantage of summarizing the benefits of treatment over a clinically relevant period, e.g. 2 h’ (43). Ramadan pointed to the common use of endpoints such as SPID in pain studies (41). The version of SPID weighted according to the time points of pain intensity assessment proved to be the endpoint with the highest responsiveness in the present study. However, SPID and %SPIDweighted are not easy to interpret. The time course of pain reduction is of higher importance for the patients in the treatment of acute pain, including headache, than for example in the treatment of chronic pain under steady state conditions of the treatment. Tfelt-Hansen et al., in their review of single attack data, found that SPID did not appear to add anything and assumed that SPID usually gives similar results to other headache relief measures (44). Our analysis using the ROC method supports this assumption. The ROC method allows the quantification and comparison of the responsiveness between clinical endpoints of very different types as the ROC curve depends only on the ranks of the observations and is independent of the scale used to measure the endpoint.
Responsiveness, the ability of an outcome measure to detect clinically important changes in a specific condition of a patient, is not yet sufficiently considered in the discussion of possible endpoints in both IHS guidelines, even though it is an aspect of great relevance in clinical trials. This should be added in future revisions of these guidelines.
Footnotes
Funding
This work was supported by Boehringer Ingelheim Pharma GmbH & Co.KG Germany.
Conflict of interests
BA and HP are employees of Boehringer Ingelheim Pharma GmbH & Co. KG, Germany. BP declares no conflicts of interest. HCD received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from: Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, Coherex, CoLucid, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Grünenthal, Janssen-Cilag, Lilly, La Roche, 3M Medica, Medtronic, Menerini, Minster, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brümmer, SanofiAventis, and Weber & Weber. HCD has no ownership interest and does not own stocks of any pharmaceutical company.