
Abstract
Background: Cortical spreading depression (CSD) has an important role in migraine with aura. Prolonged neuronal depression is followed by a late excitatory synaptic plasticity after CSD.
Method: Intra- and extracellular recordings were performed to investigate the effect of CSD on intracellular properties of mouse neocortical tissues in the late excitatory period.
Results: During CSD, changes in the membrane potentials usually began with a relatively short hyperpolarization followed by an abrupt depolarization. These changes occurred roughly at the same time point after CSD as the beginning of the negative extracellular deflection. Forty-five minutes after CSD, neurons showed significantly smaller amplitude of afterhyperpolarization and a reduced input resistance. Depolarization and hyperpolarization of the cells by constant intracellular current injections in this period significantly changed the frequency of the action potentials.
Conclusion: These data indicate higher excitability of the neocortical neurons after CSD, which can be assumed to contribute to hyperexcitability of neocortical tissues in patients suffering from migraine.
Introduction
Cortical spreading depression (CSD) is a pronounced self-propagating depolarization of neurons and glia. CSD spreads slowly from the site of onset as a radial wave across the neuronal tissue with a velocity of 2-5 mm/min (1). CSD waves are associated with dramatic failure of brain ion homeostasis, efflux of excitatory amino acids from nerve cells, increased energy metabolism, and changes in cerebral blood flow (2). The hypothesis that migraine aura is the human equivalent of CSD is well established (3). CSD also occurred following head injury or intracranial haemorrhage in human neocortex (4). Furthermore, experimental investigations indicated that spreading depression (SD) may have a role in epilepsy, spinal cord disorders, and transient global amnesia (5,6).
A brief period of neuronal excitation heralds CSD, which is immediately followed by a prolonged nerve cell depression (2). However, recent studies revealed a late excitatory period following the depression phase of CSD. This late excitatory period may have an important role in the pathophysiology of CSD-related neurological disorders (7). It has been shown that induction of CSD initially suppresses synaptic activity, which is followed by an irreversible potentiation of synaptic plasticity in human neocortical tissues obtained during epilepsy surgery (7). Using quantitative receptor autoradiography, an enhancement of glutamate N-methyl-
Intracellular characteristics of cortical neurons during CSD were analysed by several investigators (12–17). It has been reported that spontaneously firing cells are inhibited from firing by hyperpolarization at the onset of a CSD and then return to the resting potential level without firing. These neurons are inactive throughout depolarization. During the early phase of depolarization, some neurons transiently increase their firing rate before inactivation (14,15,17). However, none of these studies investigated the intracellular activities in the late excitatory phase of CSD, about 45 min after initiation of negative direct current (DC) deflections. The present observations were aimed to analyse intracellular neuronal activities in the late excitatory phase of a CSD in the neocortex of adult mice.
Materials and methods
The experiments were carried out on neocortical slices of mice (Black 6, 25–30 g, 9-11 weeks old). Mice were killed by decapitation. The brain was removed under deep methohexital anaesthesia. The cerebellum was removed and a cut was made to divide the two cerebral hemispheres. Combined amygdala–hippocampus–cortex slices containing the temporal cortex, the perirhinal cortex, the entorhinal cortex, the subiculum, the dentate gyrus and the hippocampus as well as the amygdala (500 µm) were cut in a nearly horizontal plane. Dissection of the desired brain areas was performed according to the mouse brain atlas (18). The slices were preincubated at 28°C for 60 min in artificial cerebrospinal fluid (ACSF). The ACSF contained (in mmol/l): NaCl 124, KCl 4, CaCl2 1.0, NaH2PO4 1.24, MgSO4 1.3, NaHCO3 26 and glucose 10. The ACSF was continuously equilibrated with 5% CO2 in O2, stabilizing the pH at 7.35–7.4. After 30 min pre-incubation, CaCl2 was elevated to 2.0 mmol/l. The slices were transferred to an interface recording chamber and superperfused with ACSF at 32°C.
Electrophysiological recordings
Intracellular recordings were performed in the fifth layer of the somatosensory neocortex using sharp microelectrodes filled with 2 mol/l potassium methylsulphate (connected to a 2 M KCl solution bridge through a ceramic junction). Two-hundred ms square positive and negative current pulses were applied to the neurons to determine the neuronal input resistance and discharge patterns. A constant positive or negative current was injected to the cells and the membrane potentials were reduced to −35 mV and elevated to −75 mV, respectively. The reference electrode and the connection to the microelectrode were symmetric Ag–AgCl bridges. The microelectrodes were selected to have a resistance 80–160 MΩ. Extracellular field potentials were recorded simultaneously using glass microelectrodes filled with 150 mmol/l NaCl and with a resistance of 2–10 MΩ in the fifth layer of somatosensory neocortical slices. The potential of the intracellular electrode was referred to an extracellular microelectrode to ensure control of the true membrane potential during large shifts of extracellular potential. Extracellular recordings were obtained using a custom-made differential amplifier and the membrane potential fluctuations were obtained using a home-made active bridge mode amplifier (19). Traces were digitized by a Digidata 1200 (Axon Instruments, CA, USA) and the data were collected and analysed by Axoscope 10 (Axon Instruments).
The amplitudes of action potentials were measured from the resting membrane potential baseline to the peak. The duration of action potentials was measured as the half-amplitude width. The amplitudes of afterhyperpolarizations were measured from resting membrane potential to peak. The neuronal input resistance was calculated by Ohm's law from the ratio of the voltage deflection to the injected current. These measurements were performed for 5-min periods, 5 min before (−5 to 0 min) and 45 min (+45 to +50 min) after CSD induction. For each minute of experiments, 20 action potentials were randomly selected. Intracellular recording data acceptable for inclusion in the study met the following criteria: recording stability without any sign of injury discharges, membrane potential more negative than −45 mV with deviation less than 5% during the control period. All data are given as mean ± SEM. The data were statistically processed and compared using paired Student’s t-test. Significance was established when the probability values were less than 0.05.
Induction of SD
A glass electrode filled with 3 M KCl or ACSF (control) was fixed in a special holder connected via a plastic tube to a pressure injector. The tip was inserted into the somatosensory neocortical slices (layer I–II). A high-pressure pulse was applied to inject an amount of K+ into the tissue sufficient to induce SD (tip diameter, 2 µm; injection pressure, 0.5–1.0 bar applied for 200–300 ms, two separate injections, 1–3 nl per pulse, 2–5 mm distant from nearest recording electrode). The same amount of ACSF was applied in the slices in control experiments. The amplitudes, duration and velocity rates of SD were evaluated. Duration of DC potential fluctuation width was measured at its half-maximal amplitude. To estimate CSD propagation velocity, the distance between the two recording electrodes was divided by the time elapsed between the CSD onsets at the first (the third cortical layer) and second (the fifth cortical layer) recording sites. The CSD delay was defined as the time interval from the KCl injection until the onset of CSD at microelectrodes located in the third layer.
Experimental protocol
The experimental protocol consisted of two periods as follows. Period i (control period): the slices were superfused with ACSF (30 min); test for spontaneously appearing CSD; field potential recording was performed in the third and fifth layers of the neocortex. Period ii (induction of CSD, injection of 3 M KCl or ACSF): intracellular recordings and field potential recordings were performed simultaneously in the fifth layer of the somatosensory neocortical slices. KCl (induction of CSD) or ACSF (control) was applied after 15 min of intracellular recordings of the membrane potential fluctuations. Intracellular recordings were continued for at least 60 min after KCl or ACSF injection.
Results
Induction of CSD
After local application of KCl, extracellular recordings first showed a slight positive shift, followed by a brief burst of high frequency spikes that heralded the appearance of the negative DC potential waves (n = 22, amplitude 15.5 ± 1.2 mV, duration 125 ± 5 s; Figure 1). The delay between KCl injection and CSD initiation was 8 ± 0.9 s. SD waves propagated inversely to the direction of the ACSF flow at a propagation velocity of 3.3 ± 0.2 mm/min. Local injection of ACSF into the neocortex did not elicit any negative DC deflection.
Membrane potential changes (Vm) of a neocortical cell (somatosensory cortex; mouse) during a cortical spreading depression (CSD) event along with extracellular field potential recording (FP) near the impaled neuron. (A) After application of KCl in layer I–II of the neocortex, neurons depolarized first gradually and slightly, then abruptly at very nearly the same point of time (∼10 s) at which the negative extracellular fluctuation started. The recovery from a CSD event was usually followed by overshooting of the control level in both intra- and extracellular recordings. Note the brief burst of high-frequency spikes recorded during an early period of the DC deflection. (B) Traces of intracellular recordings 5 min before and 45 min after induction of CSD. Negative voltage is represented by a downward deflection in extracellular FP recording.
The effect of CSD on intracellular activity
Characteristics of membrane potential changes in somatosensory neocortical slices before and after induction of cortical spreading depression

Characteristics of membrane potential changes in the fifth layer of somatosensory neocortical slices 5 min before and 45 min after induction of cortical spreading depression (CSD) at the resting membrane potential (RMP) and after injection of a constant positive or negative current to depolarize or hyperpolarize the membrane. Note the reduction in the amplitude of afterhyperpolarization (AHP) and the neuronal input resistance after propagation of CSD. In addition, the frequency of spontaneous action potentials and the duration of action potentials were significantly reduced by CSD propagation, when the RMP hyperpolarized to −75 mV. APs, action potentials; DAPs, depolarization before action potentials. Values represent mean ± SEM. *p <0.001, **p <0.009, ***p <0.013.
During CSD events, the membrane potentials changed drastically. After application of KCl in layer I–II of the neocortex, changes in the membrane potentials usually began with a relatively short and small hyperpolarization, followed by a depolarization. Neocortical neurons depolarized first gradually and slightly, indicating a small inward membrane current. Then the neurons depolarized abruptly at roughly the same time at which the negative extracellular deflection started. At the peak of CSD (extracellular negative DC shift), the mean RMP reached −6.0 ± 2.6 mV. Depolarization of the membrane potential was followed by a hyperpolarization of smaller amplitude. The recovery from a CSD event was usually followed by overshooting of the control level by both intra- and extracellular recordings (Figure 1). APs reappeared within 6.0 ± 1.6 min after the negative DC shift returned to the baseline (recovery to baseline).
The above-mentioned parameters (APs, RMP, AHP and DAPs) were not changed for at least 90 min after injection of ACSF into the slices (n = 8). However, application of KCl and induction of CSD changed the intracellular properties of these cells (n = 22). Forty-five min after induction of CSD by KCl application, RMP, DAPs, the mean duration and the amplitude of APs, input resistance, and the duration and amplitude of AHP were analysed (Table 1). Propagation of CSD significantly reduced the amplitude of AHP to 2.5 ± 0.3 mV compared with the control period (3.8 ± 0.5 mV; p = 0.009). Propagation of CSD also significantly decreased the neuronal membrane input resistance from 112 ± 14 MΩ (pre-CSD level) to 78 ± 9 MΩ (p < 0.001). The spontaneous firing rate did not change after propagation of SD (387 ± 83 per min) compared with the pre-CSD level (395 ± 73 per min). The APs, depolarized to −40 mV, had a significantly lower AHP in neurons affected by CSD (p <0.001). In addition, hyperpolarization of neurons to −75 mV 45 min after induction of CSD led to significantly prolonged APs (p <0.001) and to a reduction in the frequency of APs (p = 0.013) and a reduction in the amplitude of the AHP (p <0.001; Figure 2). Before induction of CSD, the current required depolarize the neurons to −40 mV and to hyperpolarize them to −75 mV were 0.49 ± 0.03 nA and 0.33 ± 0.05 nA, respectively. After propagation of CSD, the current required to change RMP to −40 and −75 was significantly increased to 0.67 ± 0.05 nA (p <0.001) and 0.51 ± 0.04 nA (p <0.016), respectively.
The membrane potential fluctuations recorded from layer V of the somatosensory neocortex before and after induction of cortical spreading depression (CSD). Intracellular injection of a constant positive or negative current was used to investigate how burst characteristics and after-potentials change at depolarized (−40 mV; B) and hyperpolarized (−75 mV; C) states of the membrane and were compared with the values observed at the resting membrane potential (A). CSD was elicited by KCl injection in layer I–II of the neocortex. Traces were selected from intracellular activities 5 min before (left) and 45 min after (right) induction of CSD. Note (i) the significant enhancement and reduction in neuronal burst firing after constant injection of positive and negative currents, respectively, and (ii) the reduction in the amplitude of the afterhyperpolarization after induction of CSD.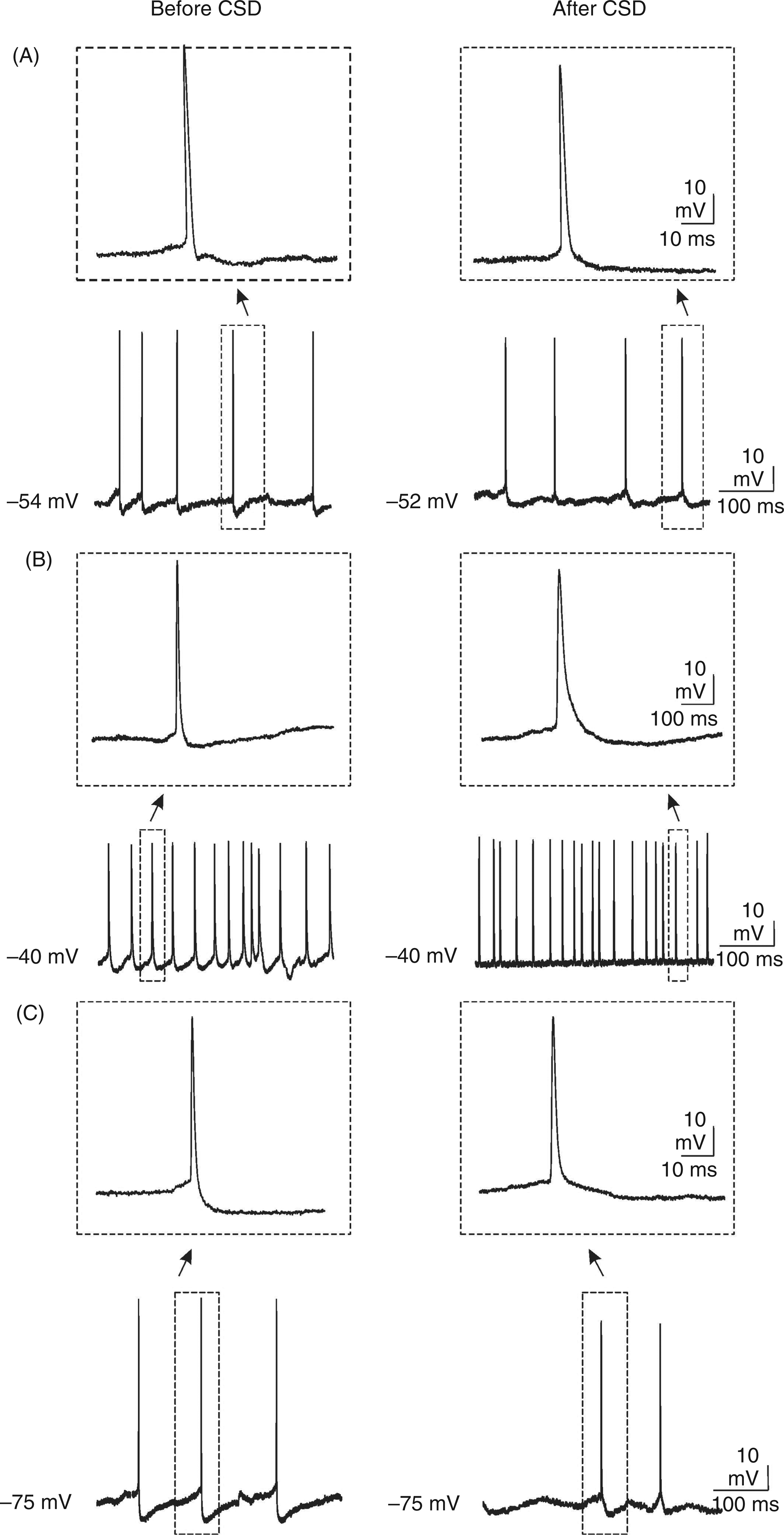
The response to intracellular current pulses injected through the microelectrode (200 ms; 100 pA) was recorded before (5 min) and after (45 min) induction of CSD. The neurons after induction of CSD displayed a higher frequency of APs (32.1 ± 0.5 vs 13.9 ± 0.4 Hz; p <0.001) with a shorter inter-spike interval (67 ± 10 vs 43 ± 10 ms; p = 0.019). In addition, these neurons also exhibited a smaller depolarization step amplitude after current injection (35 ± 8 vs 12 ± 2 mV; p <0.001; Figure 3). All above-mentioned parameters were analysed 90 min after induction of CSD (n = 12); there were no changes observed in these parameters compared with those recorded 45 min after CSD induction.
Typical discharge patterns of cortical neurons induced by intracellular injection of square positive current pulses (200 ms; 100 pA). Current injection was performed before (A) and after (B) induction of cortical spreading depression (CSD). Note (i) the higher frequency of action potentials with shorter inter-spike interval and (ii) the smaller depolarization after current injection 45 min after induction of CSD.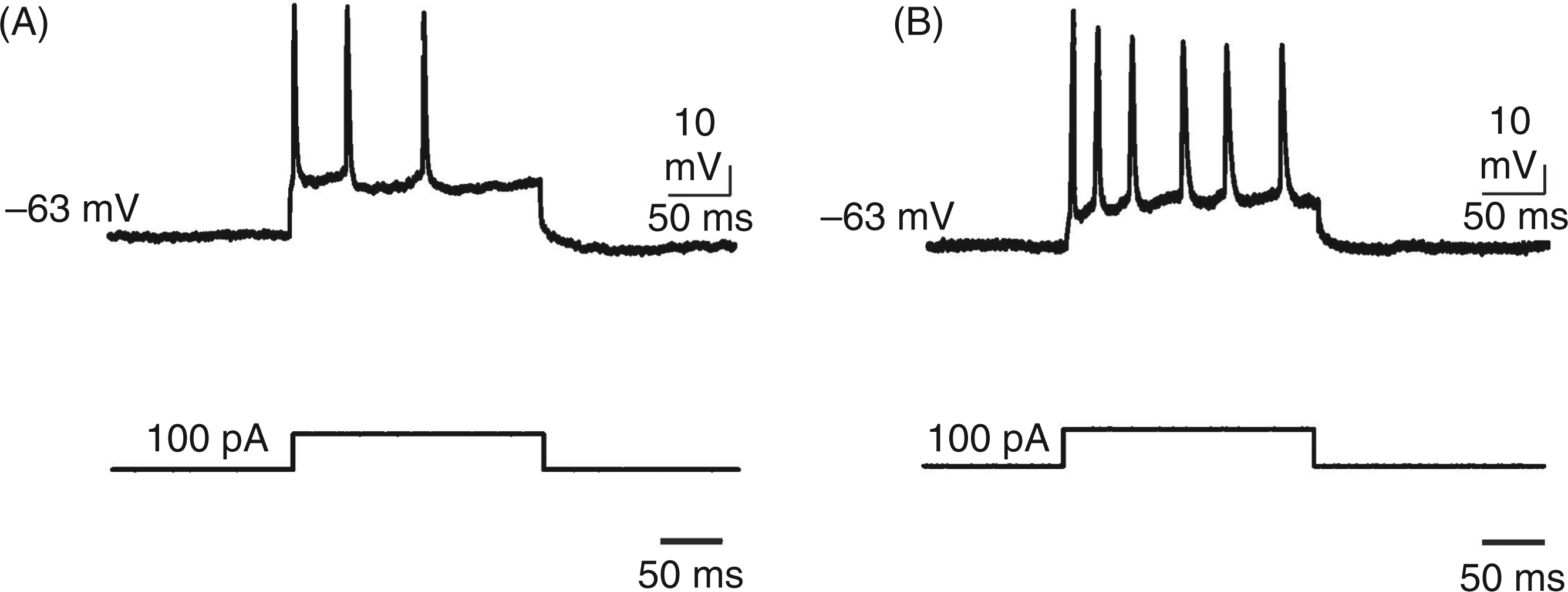
Discussion
The present data add to our understanding of the late neuronal hyperexcitability period after propagation of CSD. After induction of CSD, neocortical neurons first showed a brief burst of high frequency spikes during a slight positive shift. This short hyperexcitability heralded the large negative sustained DC potential shift accompanied by a marked depolarization of the cell membrane (transient suppressive period). After recovery from the suppressive phase, neocortical neurons are more excitable than during the control period (before induction of CSD).
Studies demonstrated the efflux of K+ from the neurons during CSD. The enhancement of extracellular K+ is accompanied by a precipitous drop in extracellular concentrations of Cl−, Na+ and Ca2+, suggesting that K+ leaving the cells is exchanged for Na+ and Ca2+ that are entering (2). When several ions flow across the membrane, fluctuation of the membrane potential is a joint function of the conductances and concentration ratios of the participating ions. Some but not all of the K+ leaving the cells flows through tetraethylammonium (TEA)-sensitive ion channels. ATP-sensitive K+ channels probably carry some of the K+ released during propagation of CSD (20). Before the arrival of the CSD, the membrane potential began to depolarize gradually and slightly, indicating a small inward membrane current. This inward current initiates brief high frequency spikes. During this brief period, the Na+ pump of neurons is stimulated by the increased extracellular K+ and pumps the Na+ into the cell (16). During CSD, the membrane potential shifted from a negative value toward zero, sometimes advancing into the positive domain. The depolarization could alter the kinetics of the voltage-sensitive channels, for example by inactivating the sodium channels, and suppress cellular activity. The drastic alterations of extracellular ion concentrations have been interpreted as signalling a reduction in transmembrane ion gradients.
Extracellular Ca2+ concentration decreases from its normal level of 1.2–1.5 mM to >0.3 mM, accompanied by a rise in intracellular Ca2+ to about 2 μM during CSD in neocortical neurons. Much of the Ca2+ entering cells was either bound or sequestered into organelles (21). Although the CSD-induced effects on intracellular Ca2+ were largely transient, the basal intracellular Ca2+ seemed to be slightly elevated following CSD (21,22). Intracellular Na+ loading seems to be a critical upstream requirement that contributes to loss of effective Ca2+ homeostasis during a phenomenon such as CSD (23), and Na+ loading before CSD could potentially underlie subsequent loss of Ca2+ homeostasis. Progressive compromise of mitochondrial function before CSD may also contribute to inability to restore neuronal Ca2+ homeostasis (24). This raises the possibility that there may be long-term consequences such as enhancement of cellular and synaptic activities after CSD (7).
The amplitude of AHP was reduced after CSD propagation, which was probably due to a decrease of outward K+ conductance. CSD induces cell swelling, which in turn is associated with the activation of potassium leak channels belonging to the 2-pore domain family (25). On the basis of increased extracellular K+ concentrations and of the activation of Ca2+-dependent K+ flow, the potential dependent K+ conductance underlying the AHP is expected to be increased. A reduction in the AHP, in turn, would be expected to increase neuronal excitability (26). Evidence for such enhanced excitability (a tendency to fire more APs after depolarizing pulses) has been found in the hippocampus (27). The same was found in our investigation. The calcium-dependent AHP has a fundamental role in the regulation of repetitive firing and spike frequency adaptation (28). Several studies attest that CSD modulates neuronal excitability (7,9), including long-term potentiation in the neocortex (7,29). This implies that a reduction in the amplitude of the AHP may serve as a general mechanism of increased excitability, which facilitates activity-dependent synaptic modifications (30). It was shown that blocking AHP currents enhances induction of long-term potentiation in the hippocampus (31).
Reduction in membrane input resistance during and after CSD propagation was reported by several investigators (2). At the height of CSD, ion gradients across membranes were lowered and neuronal membrane resistance was severely reduced, and gradually recovered thereafter. However, the input resistance was still significantly lower than during the control period 45 min after CSD. Partial recovery of neuronal membrane resistance after CSD has been reported (32). This can be achieved by subsequent opening of one or several of the mentioned channel candidates, well after the major ion changes that occur at the wavefront of CSD. For instance, an intracellular increase in Ca2+ could serve as an intracellular signal triggering the opening and modulation of certain channels via second messengers (33,34). The observed reduced AHP after CSD can also be explained as secondary to conditioning-induced alteration of input resistance. The AHP response measured after an action potential caused by the injection of positive current is primarily caused by a Ca2+-mediated K+ conductance (35). CSD especially reduced the early phase of AHP. This may be resulted by modulation of the maxi Kca (BK) channels (36). It has been shown that activation of BK channels reduces hippocampal susceptibility to hypoxia-induced SD (37). Reduction in input membrane resistance explains the smaller depolarization step after current injection needed to evoke burst firing after CSD in our experiments.
In line with our results, a transient depression of neuronal activities during CSD, which was replaced by hyperexcitability after CSD, was also observed in rat spinal cord tissues after propagation of CSD (6). A similar pattern of an initial inhibitory effect (during migraine attacks) and subsequent excitatory effect (1–2 h after attacks) was observed in visually evoked potentials in migraineurs (38). Migraine attacks are characterized by hypersensitivity to visual, auditory and olfactory stimuli (39). Photophobia is an abnormal sensitivity to light experienced by migraineurs during attacks. It has been reported that photophobia in patients suffering from migraine is linked with a visual cortex hyperexcitability (40). A CSD-like wave occurred during the aura phase of migraine attacks in the human brain and was accompanied by a visual aura (41). Using direct current magnetoencephalography, multiple areas of hyperexcitability in the primary visual cortex and occipital parietal regions were observed during the aura phase of migraine attacks (42). In addition, electrophysiological studies using multimodal evoked and event-related potentials found an increased cortical excitability also in the headache-free interval in migraineurs (43). Hyperexcitability of the extrastriate cortex was reported in patients suffering from migraine (44).
Impaired habituation is probably a neurophysiological marker of migraine (45). Several lines of evidence support the role of reduced neocortical inhibition in the pathophysiology of migraine (46). CSD causes a selective reduction in cortical inhibition by suppression of GABAergic synaptic plasticity (47). A hyperexcitable state in the subcortical regions in patients with migraine, during both interictal and ictal periods, was reported (48). Excitation of different subcortical regions was observed after propagation of CSD (9,10). Changes in cortical and subcortical plasticity, as well as neuronal activities, induced by CSD can be assumed to contribute to affect excitability and responsivity of the brain in patients suffering from migraine.
Footnotes
Acknowledgements
We thank Philippe Coulon for critical comments on an earlier version of the manuscript.
Funding
This study was supported by German Research Foundation (DFG/SFB-TR3-D10 to AG) and Thesis 90-03-87-14593.