
Abstract
Background: During two decades of migraine provocation studies with naturally occurring signalling molecules, vasodilators such as prostaglandin E2, prostaglandin I2 (prostacyclin) and prostaglandin D2 were shown to be able to induce headache in man. To elucidate the role of inflammation and vasodilatation in the generation of headache, we investigated whether the pro-inflammatory and vasoconstricting prostanoid prostaglandin F2α (PGF2α) would cause headache in a human model of headache.
Methods: Twelve healthy volunteers were randomly allocated to receive 3.5 µg/kg/min PGF2α or placebo over 20 min in a two-way crossover study. We recorded headache intensity on a verbal rating scale, middle cerebral artery blood flow velocity (VMCA) and the diameters of the superficial temporal artery (STA) and radial artery (RA).
Results: We found no difference in the area under the curve (AUC) for immediate headache (0–90 min) between PGF2α and placebo (p = 0.144). The McNemar's test showed no difference in the incidence of immediate and delayed headache between verum and placebo (p = 0.500 and p = 1.000, respectively). There was no difference in VMCA (p = 0.776) and in the diameter of the STA (p = 0.460) or RA (p = 0.780) between PGF2α and placebo.
Conclusion: The present study shows that PGF2α, unlike vasodilating prostaglandins, does not provoke headache. We suggest that the vasodilating abilities of prostaglandins are important for the induction of experimental headache in healthy volunteers.
Introduction
Two highly discussed topics in migraine research today are the role of inflammation and vasodilatation. Non-steroidal anti-inflammatory substances are effective in the treatment of acute migraine (1), as well as in the prophylaxis of migraine (2), and a whole avenue of research has focused on neurogenic inflammation in migraine (3). Vasodilatation has a traditional role in migraine, but in recent years increasing evidence has suggested that it may not be as important as initially thought. Strongest among such evidence is the fact that calcitonin gene-related peptide (CGRP) receptor antagonists (4) and 5-hydroxytryptamine receptor 1F (5HT1F) agonists (5) are effective in the acute treatment of migraine but have no vasoconstrictive action. We still favour that vasodilatation has some role in migraine pain because during two decades of migraine provocation studies with naturally occurring signalling molecules only vasodilators were able to induce migraine (6–8). In addition, the intravenous administration of norepinephrine (noradrenaline), well known for its vasoconstricting effect, did not cause headache or vascular responses in healthy volunteers (9).
Over the past 5 years we have systematically investigated the role of prostaglandins (PGs) in the generation of headache in healthy volunteers and migraine sufferers (8,10–12). The intravenous administration of vasodilative PGs, such as PGI2 (prostacyclin), PGE2 and PGD2 induced headache in healthy volunteers and PGI2 triggered migraine-like attacks in migraine sufferers (8,10–12). To what extent the dilatation of cerebral or extra-cerebral arteries contributes to PG-induced headache or migraine is still unknown. One might argue that PGs are inflammatory mediators (13) capable of activating sensory afferents and that PG-induced vasodilatation does not contribute to provoked headache. To elucidate the role of inflammation and vasodilatation we examined the effects of an infusion of PGF2α, a product of arachidonic acid, which is involved in tissue inflammation (13) and pain processing (14). In contrast to the other PGs, PGF2α acts as a vasoconstrictor (15). Elevated levels of urinary PGF2α metabolite in children with migraine (16) and, more recently, increased ictal levels in the saliva of migraine sufferers have been reported (17). We tested the hypothesis that the pro-inflammatory and vasoconstricting prostanoid PGF2α could cause headache.
Methods
Participants and study design
The study was designed as a randomised, double-blind, placebo-controlled, two-way intraindividual crossover study. Trial subjects were recruited through the www.forsogsperson.dk website (18), through the MOK.dk magazine for medical students of Copenhagen University, and by advertising on the bulletin boards of hospitals and higher education institutions. Twelve healthy volunteers (five male and seven female), mean age 24.4 years (range: 19–33 years) and mean weight 66 kg (range: 55–90 kg) completed the study. Participants had no history of migraine or any other type of headache (except for episodic tension-type headache less than once a month) and no previous serious somatic, psychiatric or infectious diseases. Physical and neurological examinations, and electrocardiography (ECG), were carried out on the day of enrolment.
The subjects were randomly assigned, by means of the www.randomization.com website (19), to receive 3.5 µg/kg/min PGF2α (Prostin® F2 alpha; Pfizer Ltd, Tadworth, UK) or placebo (saline) over 20 min and crossed over to receive the other alternative treatment after a period of at least 1 week. The randomisation and preparation of the study drugs was carried out by medical staff not involved in the study. The randomisation code remained in Glostrup Hospital during the study and was not made available to the investigators until the study was completed. Before the main experiment we conducted a pilot study. Four different doses of PGF2α (2 µg/kg/min, 2.5 µg/kg/min, 3 µg/kg/min and 3.5 µg/kg/min) were tested to determine the highest well-tolerated dose. The average maximum mean arterial pressure (MAP) increase during the 20-min injection of 2 µg/kg/min PGF2α was 80 (60–89 safety limits, defined in the protocol). For the 2.5 µg/kg/min PGF2α dose it was 82 (62–94), for the 3 µg/kg/min PGF2α dose it was 88 (60–90) and for the 3.5 µg/kg/min PGF2α dose it was 89 (61–91). We found that the maximal dose of 3.5 µg/kg/min PGF2α was well tolerated and, therefore. it was used in the main experiment.
The study protocol was formally approved by the Ethics Committee of the City of Copenhagen (ref VEK H-B-2008-025) and by the Danish Data Protection Agency and was performed in accordance with the Declaration of Helsinki, as revised in 2008. The study was registered on the ClinicalTrials.gov website (www.clinicaltrials.gov) (20). All subjects gave written informed consent to participate in the study. The trial was conducted according to the protocol and good clinical practice.
Headache intensity and adverse events
To record headache intensity, a 10-point verbal rating scale (VRS) was used, where 0 indicated no headache; 1 indicated a different sensation, pounding or throbbing, but not necessarily painful; 5 indicated moderate headache; and 10 indicated the worst imaginable headache (21). Participants were encouraged to self-report any changes in their well-being during the study. Subjects were questioned about the presence of adverse events (AEs), headache and accompanying symptoms, according to the 2nd edition of the International Headache Classification (ICHD-2) (22), at T-10, T0 and then every 10 min until T90. During the out-of-hospital period, defined as the period after discharge and until bedtime, all subjects were carefully instructed to make hourly recordings of headache and accompanying symptoms according to the ICHD-2 (22) and any other AEs. The AEs were classified as related to or not related to the study drug by the investigator. Subjects were allowed to take an analgesic medication of their own choice after consulting the study physician.
Transcranial Doppler ultrasonography and C-series ultrasound scan
Middle cerebral artery blood flow velocity (VMCA) was recorded using transcranial Doppler ultrasonography (2 MHz) by means of handheld probes (Multi-Dop X digital; Compumedics DWL, Sipplingen, Germany) (8,10,11). The recordings were performed bilaterally and simultaneously and measurements of the partial pressure of end-tidal carbon dioxide (PetCO2) were obtained with an open mask without any respiratory resistance (Propaq Encore®; Welch Allyn Protocol, Beaverton, Oregon, USA), as previously described (23). A fixed point was used with the best possible signal along the middle cerebral artery (MCA), as close as possible to the bifurcation of the anterior cerebral artery and MCA. The fix point was marked and noted and was reused in each participant for all recordings. All measurements were obtained by the same skilled laboratory technician.
A high-resolution, C-series ultrasound scanner (20 MHz, bandwidth 15 MHz; DermaScan C, Cortex Technology ApS, Hadsund, Denmark) was used to measure the diameter of the frontal branch of the left superficial temporal artery (STA) and the left radial artery (RA). All C-series scans were performed on the same location, as delimited by markings drawn on the skin. The coordinates of the marks were kept for reuse in the following trial days. All measurements within the same study subject were performed by the same skilled study personnel.
Experimental procedure
Participants had to completely avoid alcoholic beverages, caffeine, cocoa, chocolate and smoking for at least 8 h before the infusion was administered. The use of pharmacological agents apart from oral contraceptives was not permitted. Subjects fasted overnight and reported to the laboratory at 08:00 and were confirmed to be headache-free. Subjects rested in the supine position throughout the study period from time -10 min (T-10) (the start of the in-hospital period) to T90 (the end of the study period, 70 min post-infusion). The procedures were performed in a quiet room at room temperature between 23.3 and 24.1°C. Two intravenous Venflon™ catheters (Becton Dickinson AB, Stockholm, Sweden) were inserted into the antecubital vein for the infusion. The subjects rested for at least 30 min before the T-10 values of VMCA, the diameters of the STA and RA, MAP, heart rate (HR), PetCO2, transcutaneous arterial oxygen saturation (SatO2), ECG, headache score and AEs were recorded.
Following the baseline measurements, subjects were randomised to either PGF2α or placebo. At T0, the infusion was initiated by a time- and volume-controlled infusion pump (B. Braun Perfusor, Melsungen, Germany). All measurements were recorded at T-10, T0 and then every 10 min until T90. MAP and HR were measured by an auto-inflatable cuff (Propaq Encore®; Welch Allyn Protocol, Beaverton, Oregon, USA). The ECG was recorded continually on paper using a Cardiofax V electrocardiograph (Nihon-Cohden Corporation, Tokyo, Japan).
Statistics
Vascular variables are presented as mean ± SD and as mean percentage from baseline. Headache scores are presented as median and quartiles. Baseline was defined as the average of T-10 and T0 before the start of the infusion. Immediate headache was defined as any headache during the in-hospital period (0–90 min) and delayed headache was defined as any headache occurring during the out-of-hospital period.
Sample size was calculated for a 90% power and a two-sided 5% significance level. Based on our previous studies with prostaglandins (8,11) we assumed the incidence of headache after PGF2α as being 100% and after placebo as being 25%. The sample size was calculated as n = 21p(1−p)/(pA−pB)2, where p = (pA + pB)/2, n = 21 × 0.625(1−0.625)/(0.75)2 = 8.75 (24). As we also wanted to find a difference in the MCA of at least 10% between two study days we conducted an explorative sample size calculation: n = 2σ2/(μ2 − μ1)2 × f (α, 1 − β) = 2 × 0.12/(0, 12) × 10.5 = 21 (σ = 10%, μ2−μ1 = 10%) (25). Therefore, 12 subjects were considered to be sufficient for a crossover study.
The data were baseline-corrected and the VMCA data were also PetCO2 corrected (26). This was followed by calculating the area under the curve (AUC) for the time period T0–T90 for VMCA, and for the headache score, MAP, HR and PetCO2, according to the trapezium rule (27).
The primary end points were the difference in incidence of headache and the AUC for headache score (AUC headache score) between PGF2α and placebo. The secondary end points were the differences in the AUC for the VMCA (AUC VMCA), STA (AUCSTA), RA (AUC RA), PetCO2 (AUC PetCO2), MAP (AUC MAP) and HR (AUC HR) between the placebo day and the active day. To test the statistical difference between the variables we applied a paired, two-way t-test for the vascular data; the Wilcoxon signed-rank test was used for the AUC headache score and the McNemar's test was used for the incidence of headache and AEs. To explore any possible changes over time for vascular variables we conducted a post hoc analysis by means of a repeated measures one-way ANOVA (which included the Dunnett's test). Five per cent (p < 0.05) was accepted as the level of significance. All analyses were performed with the PASW Statistics 18 for Windows software (SPSS Inc., Chicago, Illinois, USA).
Results
We recruited a total of 16 healthy volunteers. During the study, four participants withdrew. Therefore, 12 subjects were retained, as planned in the study protocol. Three of the four participants who withdrew wished to abort the infusion on either the first or second study day due to severe nausea or increased intestinal motility and discomfort. The infusion was stopped at approximately T10 on the active day. By this time none of the participants had experienced headache. One participant reported headache on the placebo day. One participant was withdrawn from the study due to an increase in MAP of 36%, which was 16% over the safety limit (20%), as defined in the protocol.
Mean baseline values (± SD) of measured variables

PGF2α: prostaglandin F2α, VMCA: middle cerebral artery blood flow velocity (average of four cycles); PetCO2: end-tidal partial pressure of carbon dioxide.
P value: difference in baseline variables between two experimental days (paired t-test).
Prostaglandin F2α-induced headache and vascular responses
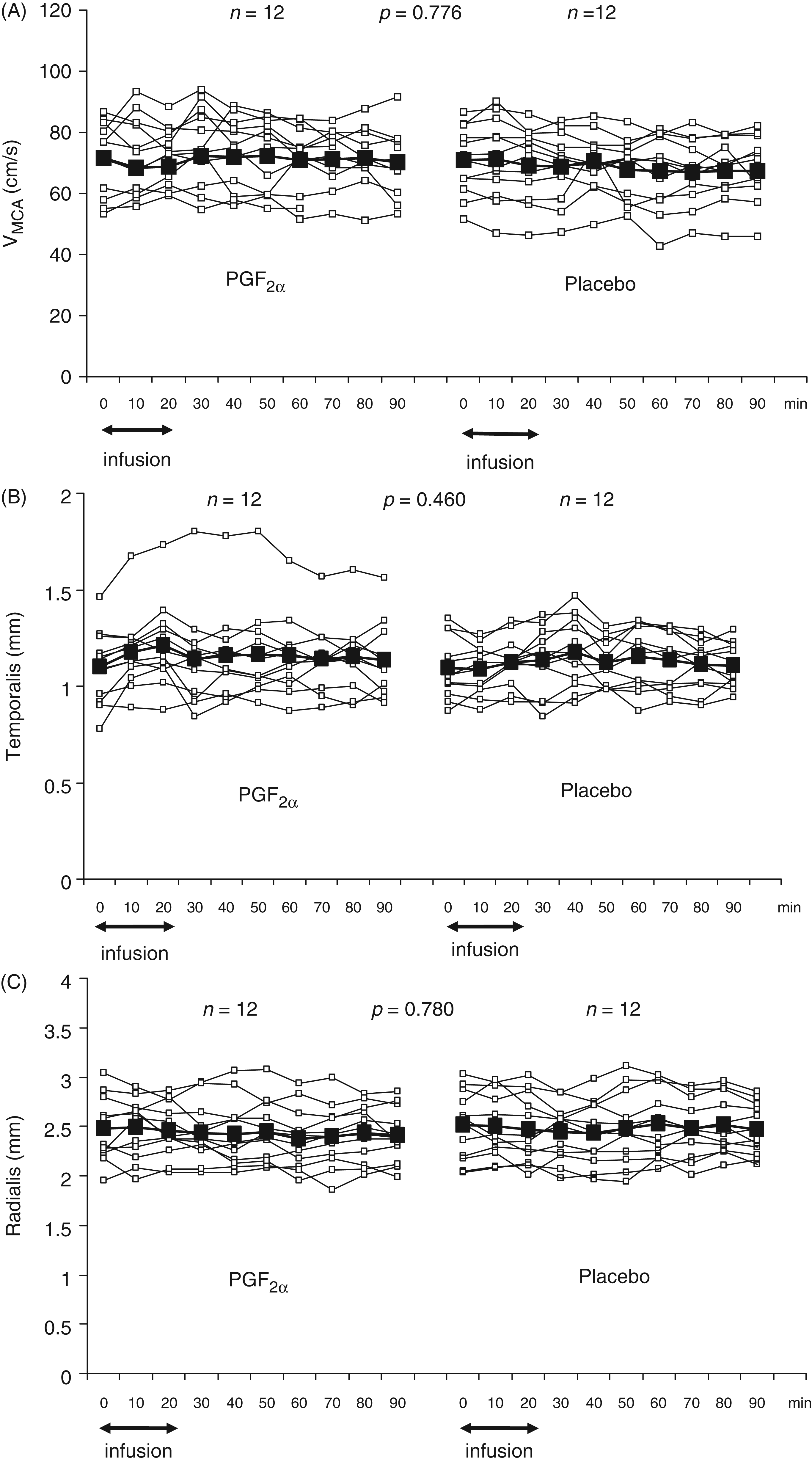
The incidence of immediate and delayed headache is shown in Table 2. In all cases of headache it was reported as pressing and localised to the forehead. We found no difference in the AUC for immediate headache (0–90 min) between PGF2α and placebo (p = 0.144) (Figure 1). There was no difference in the AUCVMCA between PGF2α and placebo (p = 0.776) (Figure 2A). Due to a difference in the AUCPetCO2 between the placebo and PGF2α day (p = 0.039), the MCA data were corrected with e0,034 for each mmHg decrease in PetCO2 (23). Exploratory ANOVA analysis revealed no changes over time in VMCA on either study day (p > 0.05). We found no difference in the diameter of the STA or RA between the PGF2α and the placebo day [AUCSTA difference (p = 0.460) and AUCRA difference (p = 0.780)] (Figure 2B and C).
Individual (□) and median (▪) headache scores on a verbal rating scale after intravenous infusion of prostaglandin F2α (PGF2α) and placebo in 12 healthy volunteers. There was no difference in the area under the curve (AUC) for headache (0–90 min) between PGF2α and placebo (p = 0.144). Individual (□) and mean (▪) (A) middle cerebral arteries blood flow velocities (cm/s) (VMCAs); (B) diameter (mm) of the superficial temporal artery (STA); (C) diameter (mm) of the radial artery (RA) after intravenous infusion of prostaglandin F2α (PGF2α) and placebo in 12 healthy volunteers. There was no difference in AUC VMCA (p = 0.776), AUCSTA (p = 0.460) and AUCRA (p = 0.780) between PGF2α and placebo. AUC: area under the curve. Incidence of prostaglandin F2α-induced immediate and delayed headache in 12 healthy subjects The McNemar's test showed no difference in the incidence of immediate and delayed headache between the placebo and active day.

Vital signs and adverse events
The AUCMAP and the AUCHR were significantly larger on the PGF2α day than on the placebo day (p < 0.0001) (Figure 3). The maximum mean increase in MAP from baseline by 18.9% [confidence interval (CI) 14.4–23.3] (Figure 4) was observed at T20 and the maximum mean increase in HR from baseline by 16% (CI 10.7–21.3) was also observed at T20 on the PGF2α day.
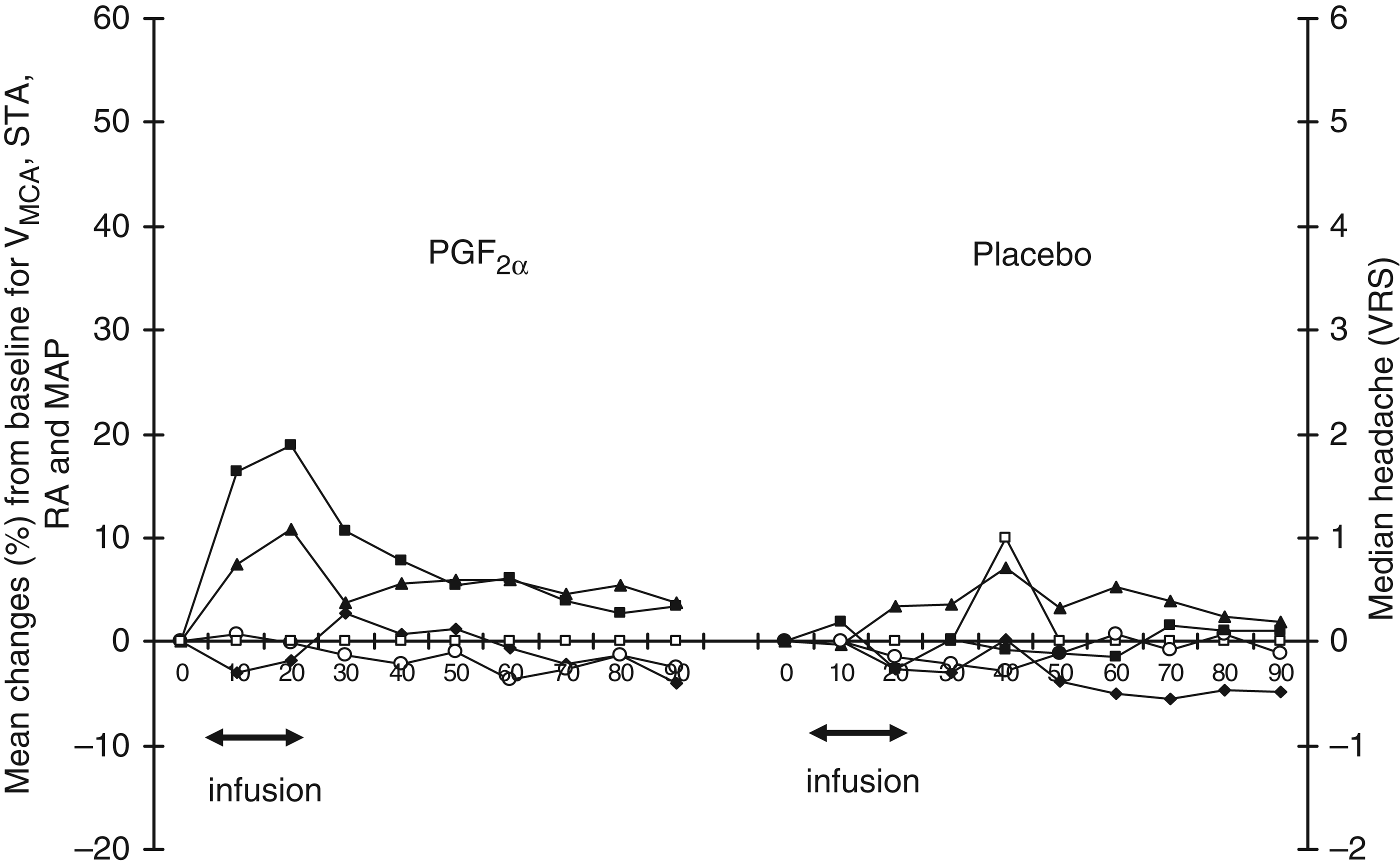
Mean MAP (▪) and HR (♦) after intravenous infusion of PGF2α and placebo in 12 healthy volunteers. The AUCs for MAP and HR were significantly larger on the PGF2α day compared with the placebo day (p < 0.0001). MAP: mean arterial pressure, HR: heart rate, PGF2α: prostaglandin F2α, AUC: area under the curve. Median headache (□) and mean percentage changes from baseline for the mean arterial blood pressure (MAP) (▪), middle cerebral artery blood flow velocity (VMCA) (⋄), diameter of the superficial temporal artery (STA) (▴) and diameter of the radial artery (RA) (♦). PGF2α: prostaglandin F2α, VRS: verbal rating scale.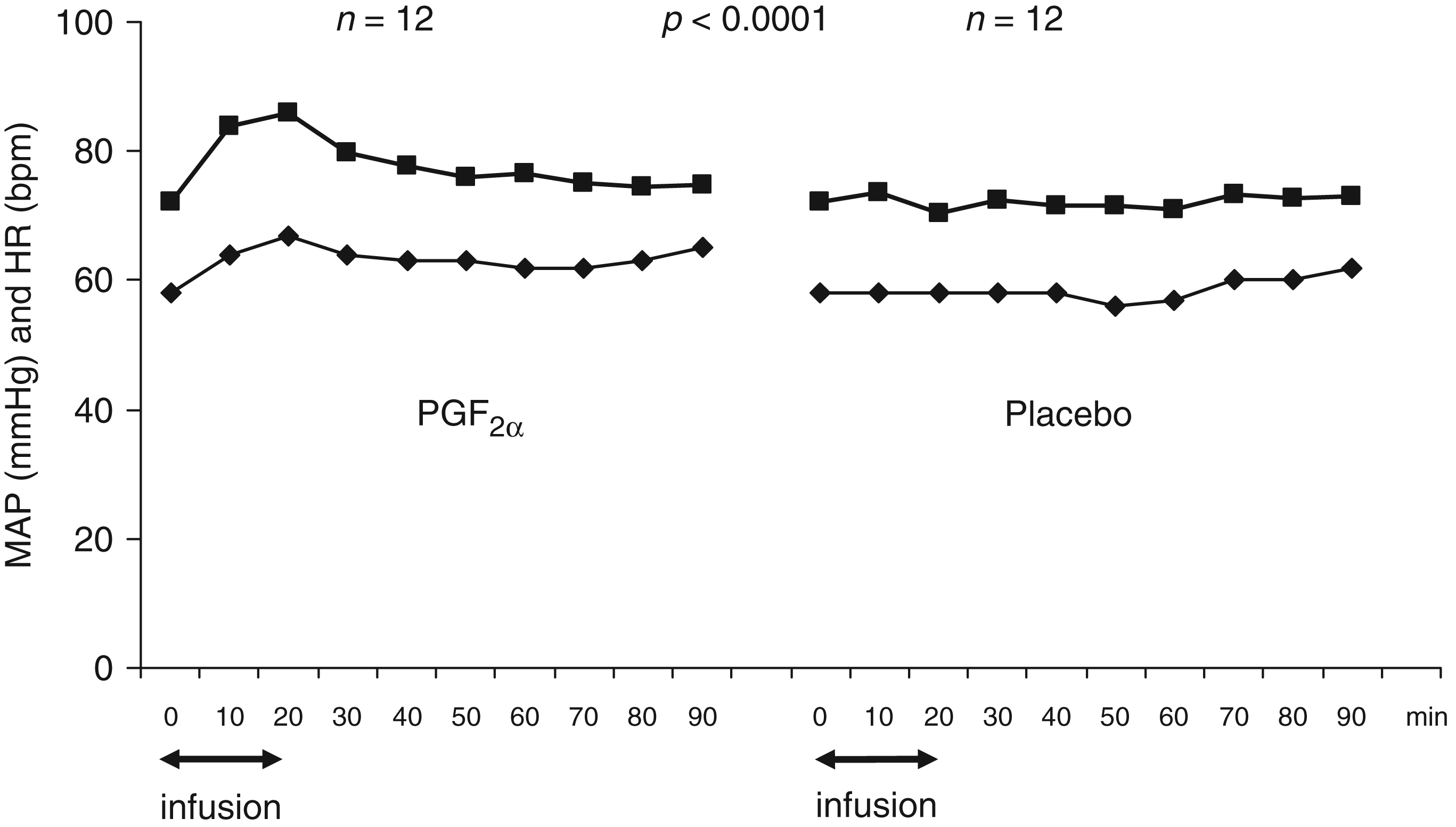
Adverse events reported and recorded during the in-hospital period (0–90 min)

PGF2α: prostaglandin F2α.
There was a difference in occurrence of heart sensation between placebo and PGF2α (p = 0.004, McNemar’s test).
There was no difference between the two experimental days regarding other adverse events (p > 0.05, McNemar's test).
Mean percentage changes of vascular parameters and median headache on the PGF2α and the placebo day are shown in Figure 4.
Discussion
The major outcome of the present study is that pro-inflammatory and vasoconstrictive PGF2α, as opposed to vasodilating prostaglandins, failed to induce headache.
PGF2α and nociception
PGF2α is one of the most abundant prostaglandins in the brain (28). Immunohistochemical studies reported the distribution of PGF synthase (PGFS) and PGF receptor (FP) in the central nervous system (29,30). PGFS immunoreactivity was found in the grey matter at all segmental levels in rat spinal cord and immunoelectron microscopy revealed increased immunoreactivity in neuronal dendrites (29). Yoshikawa et al. reported increased PGFS immunoreactivity in the white matter of the brain and spinal cord of mice (30). Furthermore, functional FP receptors were found in the spinal cord of mice (31) and mRNA for the FP receptor was expressed in cultured rat astrocytes and oligodendrocytes (32).
Intrathecal administration of PGF2α caused touch-evoked allodynia in mice (33–35) and dose-dependent mechanical hyperalgesia in rats (36). PGF2α-induced mechanical allodynia was attenuated in mice lacking the FP receptor (FP-/-) (37). Electrophysiological experiments showed that PGF2α-induced mechanical hyperalgesia corresponded well with a dose-dependent increases in mechanically evoked discharges of spinal nociceptive-specific neurons (36). Based on these data and on the ability of PGs to cross the blood–brain barrier (38), we expected that PGF2α, as a pro-inflammatory mediator, could induce headache in healthy volunteers. However, the data collected in our study showed that PGF2α did not cause headache. In the next section we discuss why PGF2α, unlike other pro-inflammatory PGs, failed to induce headache.
Why did PGF2α fail to induce headache?
Incidence of immediate headache during infusion of PGE2, PGI2, PGD2 and PGF2α and maximum per cent changes in VMCA and the diameter of the STA

PGE2: prostaglandin E2, PGI2: prostaglandin I2 (prostacyclin), PGD2: prostaglandin D2, PGF2α: prostaglandin F2α, VMCA: middle cerebral artery blood flow velocity, STA: superficial temporal artery.
Significant from the placebo day. bNot significant from the placebo day.
PGF2α-induced vascular responses in humans
PGF2α constricted isolated canine basilar, middle cerebral (44) and posterior communicating arteries (45), as well as the basilar artery in the rhesus monkey (46) and bovine MCA (47). Several human in vitro studies also reported PGF2α-induced contraction of the middle cerebral and basilar arteries of infants (gestational age: 30–40 weeks) (48) and PGF2α-induced contraction of adult pial (49), temporal (50) and radial arteries (51,52). The majority of studies which used in vivo cranial window models in rats and cats showed constriction of the basilar (53) and pial arteries (54,55) after extraluminal application of PGF2α. Interestingly, one open cranial window study in cats demonstrated dilatation of small and large feline pial arterioles during topical application of PGF2α (56). Another study which used a closed cranial window model demonstrated no response from the same vessels (57). In contrast to preclinical studies, the present study of intravenous PGF2α in human volunteers did not show constriction of the investigated brain arteries. Since we observed a significant increase in blood pressure during PGF2α infusion, a specific immediate constriction of arterioles must have been induced by PGF2α. This is in agreement with a previous study in FP (−/−) mice which showed abrogation of an immediate, dose-dependent increase in blood pressure during PGF2α infusion (58). Although there are some in vitro human studies showing constriction of both cerebral and extra-cerebral arteries in the presence of PGF2α (48–52), little is known about FP receptor distribution in human intra- and extra-cranial arteries. Our finding of a lack of arterial constriction after PGF2α administration might be explained by species-specific responses to PGF2α exposure in vivo or possibly to a simultaneous activation of other dilating prostanoid receptors. Thus, a crosstalk between FP and PGE2 (EP2) receptors could result in an increase of cyclic AMP (cAMP) (59) and an attenuation or decrease of vasoconstriction caused by the FP receptor.
Conclusion
The present study demonstrates that PGF2α, as opposed to PGE2, PGI2 and PGD2, does not induce headache. The apparent lack of a dilating effect of PGF2α on cerebral arteries could explain the absence of headache. Based on these data we suggest that the vasodilating abilities of PGs are important in generating pain in healthy volunteers.
Footnotes
Acknowledgements
We thank Lene Elkjær and Winnie Grønning for their excellent technical support.