
Abstract
Background: We investigated efficacy and tolerability of two tablets of the fixed combination of 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine (Thomapyrin) in comparison to two tablets of placebo in a post-hoc analysis of a subgroup of patients with severe headache.
Methods: Patients where included if they were used to treating their episodic tension-type headache or migraine attacks with non-prescription analgesics and reported a history of headache attacks characterized by at least severe pain and greatly impaired usual daily activities and treated headaches with pain intensity of at least 48 mm assessed on a 100-mm visual analogue scale and associated with greatly impaired usual daily activities.
Results: For the primary endpoint ‘time to 50% pain relief’ in this intention-to-treat subset (n = 179 patients), the fixed combination of ASA, paracetamol, and caffeine was statistically significantly superior to placebo (p = 0.0008). The superior efficacy of the triple combination could also be shown for all secondary endpoints such as time until reduction of pain intensity to 10 mm, weighted sum of pain intensity difference (%SPIDweighted), extent of impairment of daily activities, and global assessment of efficacy. Both treatments were well tolerated. The incidence of adverse events observed was low. The results for this subgroup analysis are consistent with respect to all endpoints and to the patients with non-severe headache and the overall patient population. As with all post-hoc subgroup analyses, the findings are hypothesis generating only and must be interpreted with caution.
Discussion: The results of this subgroup analysis confirm that the fixed combination of ASA (250 mg), paracetamol (200 mg), and caffeine (50 mg) is effective and well tolerated in a broad spectrum from mild to severe migraine and tension-type headache severity independently of the headache diagnosis.
Keywords
Introduction
Migraine with and without aura as well as tension-type headache, as defined by the International Headache Society (1), are very common diseases all over the world. In Europe, the 1-year prevalence of migraine in adults is 8% among men and 18% among women (2). The overwhelming majority of headache patients use medication for their headache (e.g. 95% of men and 97% of women with migraine). About two-thirds of patients with migraine (3) and more than 80% of tension-type headache patients (4) have never consulted a physician for their headaches, and use over-the-counter (OTC) drugs for headache treatment.
In Germany, the 1-year prevalence for all headache disorders was 60.2%, for total migraine 10.6%, migraine with visual aura 3.6%, and severe non-migrainous headache 24.7% (women 66.6%, 15.6%, 5.6%, and 27.1%; men 53.0%, 5.3%, 1.5%, and 22.2%, respectively) (5). Severe headaches were reported by approximately 60% of headache sufferers, 30% of which were migraine attacks. Women suffered significantly more frequently from severe and frequent headaches, reported disability significantly more often, and rated their health worse than men. Of the 60% of sufferers, 14.6% describe their headaches as mostly severe, 24.4% as mostly light, and the remaining 21.1% as sometimes severe and sometimes mild (5).
Over half of headache sufferers (53%; women 57.4%, men 45.5%), making up 24.5% of the general population, report having treated their headache with analgesics in the last 4 weeks. This figure rises to 81.9% of all sufferers referring to the last year (women 84.3%, men 77.8%). Predominantly, OTC medications were used to this aim (in the last 4 weeks 39.5%), while prescription preparations were used by 8.0%, and 5.5% used both prescription and OTC pain medication (5).
The single or combination preparations recommended by the medical societies for self-medication of headache have shown efficacy in randomized, controlled clinical studies (6). The fixed-dose combination of acetylsalicylic acid (ASA), paracetamol, and caffeine is one of the most frequently used OTC analgesics. In a large clinical trial, this combination was found to be statistically significantly superior to the combination without caffeine, the mono-substances ASA, paracetamol, caffeine, or placebo (7). Patients who treat their headaches themselves with OTC analgesics have mostly not received a medical diagnosis or therapy advice. Considering the large number of patients who suffer from severe headaches, the question examined in this publication, namely the extent to which findings obtained for this study population in general also apply to the subgroup of patients with ‘severe headache’, is of particular interest.
Methods
Patients
In a multicentre study, conducted between September 1998 and January 2003 and reported in details previously (7), male or female outpatients (18–65 years) were enrolled by practitioners and specialists in general and internal medicine throughout Germany. Patients who were not consulting for headache were asked whether they had headaches which they treated with non-prescription analgesics. Usual headaches had to meet International Headache Society (8) criteria for episodic tension-type headache (2.1) and/or migraine with or without aura (1.1, 1.2.1). Patients must have experienced these headaches for at least 12 months with a minimum of two headache episodes within the previous 3 months. Patients meeting at least one of the following criteria were excluded: patient treats their headache with prescription analgesics or migraine drugs, patient requires higher single doses of non-prescription analgesics to treat their headache than indicated in the patient information leaflet, patient normally treats their headache with non-prescription analgesics in effervescent tablet form, headache occurs on more than 10 days per month or lasts untreated normally less than 4 h, and close association between the occurrence of headache and menstruation (menstrual migraine). Further exclusion criteria were: concomitant treatment with prescription-only and/or non-prescription analgesics, antidepressants or antipsychotic medication (within the previous 4 weeks before study enrolment), anti-rheumatic or anti-inflammatory drugs that may influence the headache symptoms (within the previous 4 days), and drugs containing ASA (above a daily dose of 100 mg/day), paracetamol, or caffeine, as well as migraine prophylaxis or administration of drugs that influence headache symptoms. Drug overuse connected with the headache (defined as the administration of analgesics or other drugs for the treatment of acute headache on more than 10 days per month) and/or alcohol or drug abuse as defined by DSM-IV were also exclusion criteria as well as pregnancy and lactation, gastrointestinal ulcers, pathologically increased bleeding tendency, glucose-6-phosphate dehydrogenase deficiency, hypersensitivity to paracetamol, caffeine, ASA, salicylates, and other anti-inflammatory/anti-rheumatic agents or other allergenic substances, bronchial asthma, concomitant treatment with anticoagulants, chronic or recurrent gastrointestinal symptoms, liver disorders, pre-existing renal damage, Gilbert’s syndrome, or hyperthyroidism. A simultaneous participation and/or participation in another clinical trial within 4 weeks of entering this study was not allowed (7).
The study was approved by local ethics committees and patients gave written informed consent. The trial complied with the FDA Guideline for the Clinical Evaluation of Analgesic Drugs (9), with the Guidelines for controlled trials of drugs in migraine and with the Guidelines for trials of drug treatments in tension-type headache of the International Headache Society (10,11).
Patients were included in this subgroup analysis only if they reported:
a history of headache attacks characterized by at least severe pain on a 5-point verbal rating scale (VRS, 0 = no pain, 1 = mild pain, 2 = moderate pain, 3 = severe pain, 4 = very severe pain); a history of at least greatly impaired usual daily activities assessed on a 4-point VRS (1 = not impaired, 2 = somewhat impaired, 3 = greatly impaired, 4 = usual daily activities impossible); treated headache episodes with pain intensity of at least 48 mm assessed on a 100-mm visual analogue scale (pain intensity) [VAS(PI)] (12); and treated headache episodes associated with greatly impaired usual daily activities.
All four criteria have to be fulfilled in order to qualify a patient for inclusion in the subgroup of patients with severe headache attacks. The lower limit of 48 mm on the VAS(PI) to define severe pain was deduced from data obtained when patients were trained on the handling of the VAS(PI) after enrolment into the study and before randomization. The investigator gave a standardized explanation of the VAS using a written explanatory text. He handed a VAS to the patient and named one of the categories of a 6-point verbal rating scale (VRS) used to describe pain intensity (0 = no pain, 1 = mild pain, 2 = moderate pain, 3 = severe pain, 4 = very severe pain, 5 = most severe pain imaginable). Within 30 seconds, the patient was to draw a vertical line on the horizontal scale, where, according to their assessment, the mentioned pain intensity would be represented, based on the experience of their own headache. This procedure was repeated until all six categories had been named randomly by the investigator according to a randomization list provided (PROC PLAN, SAS Institute Inc., Cary, NC, USA). Cut-off points were determined at which the categories of the VRS could best be mapped to the VAS(PI). These cut-off points were deduced from receiver operating characteristic (ROC) curves (13). Various cut-off points on the VAS were regarded where patients irrespectively of the actual VRS were assigned to the higher pain category if the mark on the VAS was above the cut-off point or to the lower pain category if the mark was bellow. If the patient was named the higher category on the VRS and their mark on the VAS was above the cut-off point, the outcome was called true higher or true positive. The rate of true positive outcomes is called the sensitivity. If the patient was named the lower category on the VRS and their mark on the VAS was below the cut-off point, the outcome was called true lower or true negative. The rate of true negative outcomes is called the specificity. The ROC curve displays the true positive (sensitivity) versus the false positive (one minus specificity) rates for the range of possible cut-off points. Finally, the cut-off point was chosen at which the sensitivity and specificity were closest assuming equal importance of sensitivity and specificity. The best fit with the VRS category ‘severe pain’ was found using the cut-off point of 48 mm on the VAS(PI).
Study design and treatments
The study was designed as a randomized, placebo-controlled, double-blind, multicentre parallel group trial. Three independent headache episodes were treated by every patient. Patients treated their first headache attack with their usual analgesic (open run-in phase) and recorded the features of the headache attack in a diary. The following two headache episodes were treated with the investigational study medication (treatment phase 1 and 2). Patients qualifying for this double-blind treatment phase were randomly allocated to one of six treatment groups for both treatment phases (7). This analysis included the subgroup of patients treated with the triple combination of ASA, paracetamol, and caffeine (ASA + PAR + CAF) or placebo (PL) based on the 4:1 (ASA + PAR + CAF:PL) allocation scheme used in the study. The randomization list was generated using the commercial program ClinPro/LBL version 6.0 (Clinical Systems Inc., Garden City, NY, USA). Eligible patients were assigned by the investigator in sequential order of entry after completion of the study prephase. Access to the randomization code was strictly controlled and the treatment assignment remained unknown to all parties involved in the trial until formal database lock.
Study medication was delivered by Boehringer Ingelheim Pharma, KG, Germany. Blinding was ensured by using matched trial supplies, identical in colour, size, shape, and taste. The trial medication was to be taken as a single dose when the headache occurred and when the patient would normally have taken their usual analgesic.
Patients’ medical histories were recorded by the investigator at the screening visit. A structured questionnaire was used for the headache diagnosis. An independent headache expert performed a blinded classification of each of the three headache episodes for all study patients using this questionnaire (7).
Patients were allowed to use rescue medication 4 h after the administration of the trial medication, if their pain remained: Time of administration, the type of drug, and the dose had to be documented by the patient in the patient diary.
Patients were instructed carefully to avoid any caffeine-containing beverages and/or food within 2 h before and within 4 h after administration of the medication. Otherwise, the type and quantity of such food or beverage had to be documented in the patient diary. Usual daily caffeine intake history was recorded at the screening visit.
Patients had to contact the investigator after each headache episode for their follow-up visit. The investigator reviewed the completed diary with the patient to ensure that all required information including safety and tolerability issues had been recorded.
Endpoints
The calculated time to 50% pain relief was chosen as primary endpoint based on the pain intensity recorded on the 100-mm VAS(PI). To this purpose, all patients recorded the pain intensity prior to and then 30 min and 1, 2, 3, and 4 h after drug intake in the diary. The time to 50% pain relief was calculated by linear interpolation between adjacent observation time points. The first time point with 50% relief was used. At baseline, the headache pain intensity had to be greater than 30 mm. Patients were asked to record date and time of administration in their patient diary to the nearest 5 min.
Secondary endpoints were:
calculated time until reduction of pain intensity to 10 mm VAS(PI); percentage of patients with 50% pain relief at least after 30 min, 1, 2, 3, and 4 h [evaluated on VAS(PI)]; pain intensity after 30 min, 1, 2, 3, and 4 h [evaluated on VAS(PI)]; %SPIDweighted [weighted sum of pain intensity difference (SPID) expressed as a percentage of the maximum achievable SPID]; extent of impairment of daily activities prior to and after 30 min, 1, 2, 3, and 4 h of drug administration (4-point VRS: ‘not impaired’, ‘somewhat impaired’, ‘greatly impaired’, ‘usual daily activities impossible’); global assessment of efficacy by the patient (4-point VRS: ‘very good’, ‘good’, ‘less good’, ‘poor’); global assessment of tolerability by the patient and investigator (4-point VRS: ‘very good’, ‘good’, ‘less good’, ‘poor’); recording of adverse events (AEs) [time of onset, duration, and intensity of AEs; the intensity was determined by subjective evaluation of the patient and classified as mild (signs or symptoms easily tolerated), moderate (discomfort sufficient to cause interference with normal activities), and severe (incapacitating with inability to do work or undertake normal activity); the investigator determined the relationship between the drug treatment and AE].
The primary and secondary endpoints were the same as used in the analysis of the main trial and were all prespecified a priori (7).
Statistical analysis
The required sample size for the initial study was estimated to be 1695 treated patients (485 patients treated with ASA + PAR + CAF and 121 patients treated with placebo), giving the study a power of at least 0.80 to detect a difference between the triple combination and all other treatment groups assuming a difference in the average hourly success rate of approximately 20–40% (7,14) related to the primary endpoint. The level of significance was set at 0.05 (two-sided).
Data missing for any scheduled efficacy evaluation was replaced by the last observation carried forward procedure. If the baseline value was missing, the first measured value was used as baseline value. For patients requiring rescue medication, post-rescue medication pain intensity assessments were assigned the last recorded value before intake of rescue medication. The least favourable category (poor) was used for the global assessment of efficacy.
The efficacy and tolerability endpoints were evaluated comparatively for the subgroup of the intention-to-treat data set. The last headache episode was the basis of the evaluation of all efficacy endpoints. If a patient only treated the first headache with the study medication, this attack was analysed.
The log rank test was used to calculate the p-value and test for statistically significant difference between the triple combination and placebo group for the primary endpoint time to 50% pain relief. The log rank test was also used to evaluate the time until reduction of pain intensity to 10 mm. An analysis of covariance model with factors centre, treatment group, and the individual baseline value as covariates was used to evaluate the pain intensity difference (PID), the same model without covariate to evaluate the endpoint %SPIDweighted. The Cochran–Mantel–Haenszel test, stratified by the baseline extent of impairment was applied to analyse the assessment of the extent of impairment of daily activities. Global assessment of efficacy and tolerability were made by means of the Wilcoxon rank sum test.
The results in the subgroup with at least severe headache and impairment were compared descriptively with the corresponding results in the complement with all patients included who did not meet the criteria for inclusion in the subgroup with at least severe headache and impairment.
Results
Patient population
The intention-to-treat data set comprised 482 patients treated with ASA + PAR + CAF and 128 patients treated with placebo (7). Of these, 137 in the ASA + PAR + CAF group and 42 in the placebo group met the criteria defined for the subgroup analysis with patients who reported a history of headache attacks characterized by at least severe pain and greatly impaired usual daily activities and treated headaches with pain intensity of at least 48 mm assessed on a 100-mm visual analogue scale and associated with greatly impaired usual daily activities (Figure 1). The group with patients who did not meet these criteria comprised 345 patients treated with ASA + PAR + CAF and 86 patients treated with placebo.
Profile of the subject disposition and inclusion in the analysis data set. ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine; ITT, intention-to-treat.
Patient characteristics
Patient characteristics and headache history – patients by subgroup with severe headache and impairment and its complement

Values are median (range) or n (%).
ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine.
Characteristics of treated headache – patients by subgroup with severe headache and impairment and its complement

Values are mean ± standard deviation or n (%).
ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine.
Efficacy results
For the primary endpoint ‘time to 50% pain relief’ with the intention-to-treat subset of patients with severe headache pain and impairment, the combination ASA + PAR + CAF was statistically significantly superior to placebo (p = 0.0008). The median time to 50% pain relief was 1 h 24 min after treatment with ASA + PAR + CAF compared with 2 h 19 min after treatment with placebo. Statistical analyses with the secondary endpoints corroborated the superiority of the combination ASA + PAR + CAF compared with placebo. The time needed to reduce pain intensity to 10 mm was statistically significantly shorter in patients taking ASA + PAR + CAF (p = 0.0008). The median time was 2 h 38 min after treatment with ASA + PAR + CAF. It lasted longer than 4 h after placebo.
Pain intensity difference relative to baseline steadily increased over time in both treatment groups after intake of the medication (Figure 2). This increase was more pronounced in patients treated with the combination ASA + PAR + CAF at all time points of pain intensity assessment after intake of the medication. A statistically significant difference was seen after 1 h (p = 0.0022), and a borderline significant difference already 30 min post medication (p = 0.0504). The statistically significantly higher reduction in pain intensity continued through the 4-h assessment interval (p < 0.0028). The resulting time interval weighted mean sum of pain intensity difference (%SPIDweighted) for ASA + PAR + CAF was highly significantly different from placebo (p = 0.0003, Table 3).
Time course of the mean pain intensity difference in the two treatment groups (intention-to-treat data set of patients with severe headache and impairment). ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine. Primary and secondary efficacy endpoints – patients in subgroup with severe headache and impairment ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine; PID, pain intensity difference.
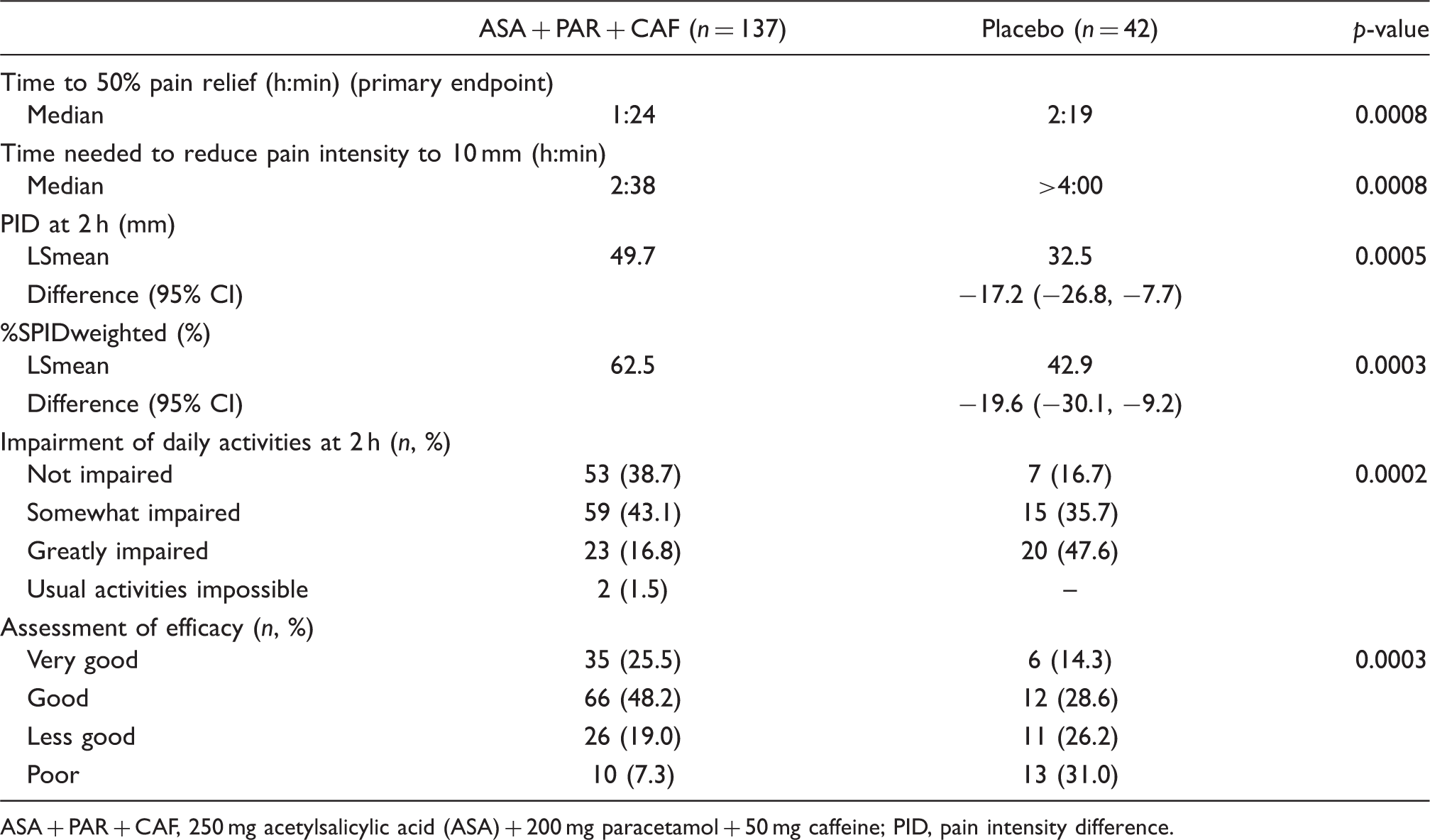
A larger proportion of patients treated with the triple combination did not experience relevant impairment of usual daily activities (Table 3). At 2 h, 81.8% of the ASA + PAR + CAF-treated patients versus 52.4% of the placebo-treated patients reported some or no impairment of usual daily activities (p = 0.0002, Table 3). The evaluation of global assessment of efficacy by the patients confirmed the superior efficacy of the combination ASA + PAR + CAF compared with placebo: 73.7% of ASA + PAR + CAF-treated patients rated the efficacy very good or good compared with 42.9% in the placebo-treated group (p = 0.0003, Table 3).
Primary and secondary efficacy endpoints – patients in complement

ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine; PID, pain intensity difference.
Assessment of efficacy missing in one patient ASA + PAR + CAF (n = 344).
Adverse events
Summary of patients with adverse events observed during randomized treatment phase – patients in subgroup with severe headache and impairment
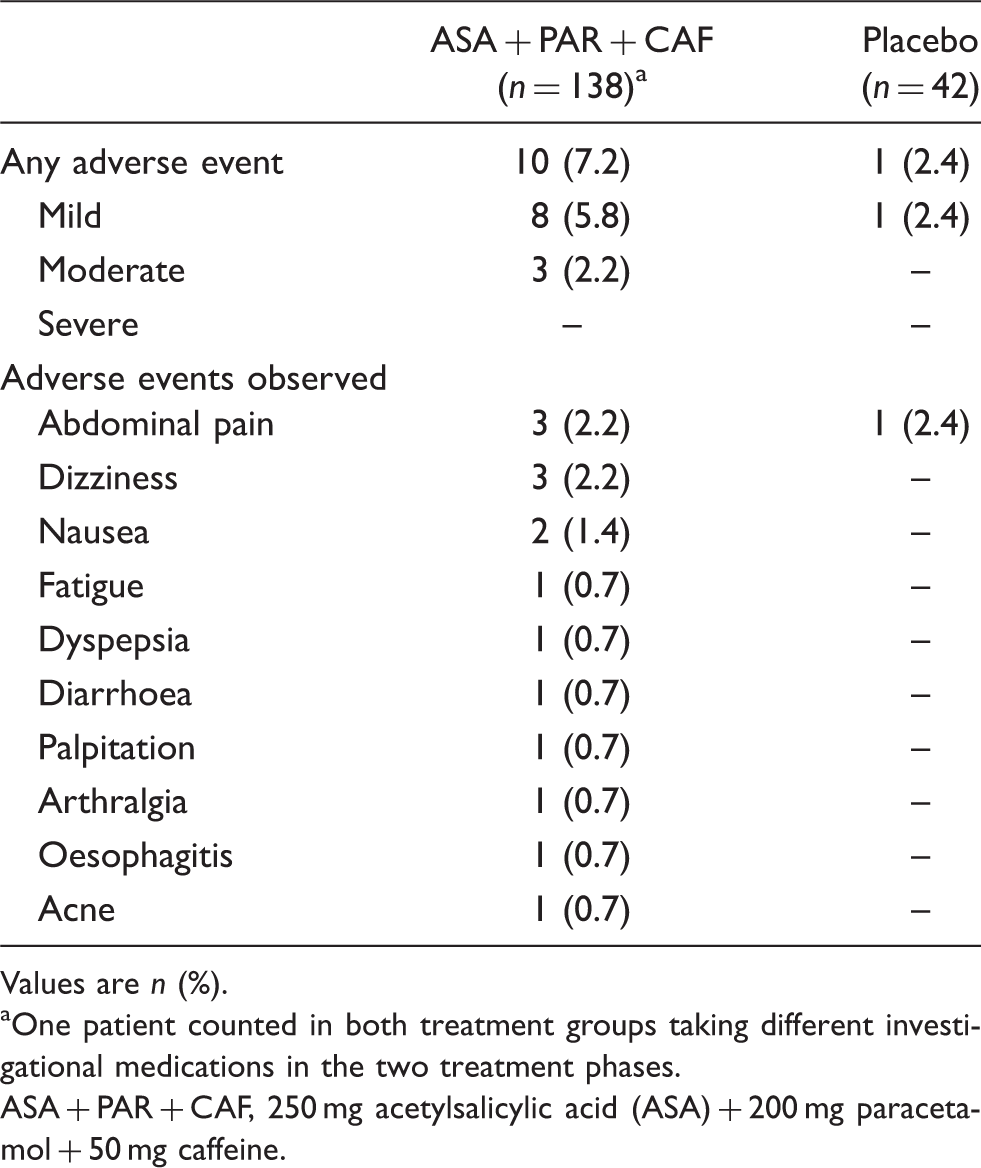
Values are n (%).
One patient counted in both treatment groups taking different investigational medications in the two treatment phases.
ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine.
Summary of patients with drug-related adverse events observed during randomized treatment phase – patients in subgroup with severe headache and impairment
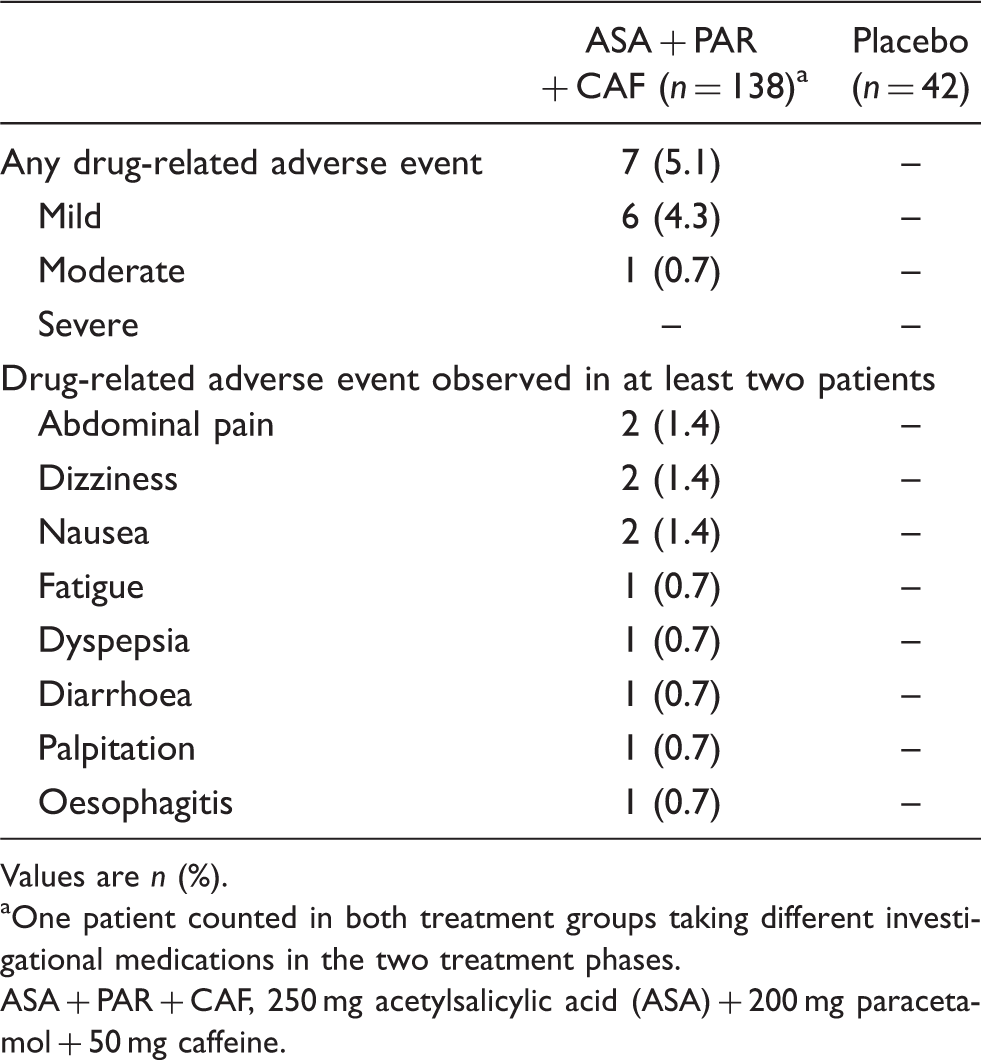
Values are n (%).
One patient counted in both treatment groups taking different investigational medications in the two treatment phases.
ASA + PAR + CAF, 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine.
Global assessment of tolerability was assessed as very good or good by the patients and investigators in more than 94% of all patients in both treatment groups.
Discussion
The results of this post-hoc analysis demonstrate that the fixed combination of ASA, paracetamol, and caffeine is effective and well tolerated in the treatment of severe migraine and tension-type headache attacks. The incidence of adverse events of this subgroup analysis is well comparable with the incidence in the overall population. For this post-hoc analysis, a very restrictive definition of the subgroup ‘patients with severe headache attacks’ was selected, which comprised four criteria: a history of headache attacks characterized by (a) at least severe pain and (b) greatly impaired usual daily activities; and with the treated headache episodes having (c) a pain intensity of at least 48 mm assessed on a 100-mm visual analogue scale and being associated with (d) greatly impaired usual daily activities. In order to ensure a clinically relevant definition of this subgroup, not only those headache episodes treated in the study were taken into account in the evaluation, but also the headache history. Those patients who were also treated with the fixed-dose combination ASA, paracetamol, and caffeine or placebo, but who did not fulfil these criteria, formed the ‘complement’ in each case. By contrast, in other studies (15), subgroups were merely defined by the intensity of the headache at baseline (pain intensity verbal rating scale ‘severe’).
Severe pain was defined in this subgroup analysis as pain that was rated at 48 mm or above on the 100-mm VAS(PI). This definition was based on data obtained during a training session of the use of the VAS(PI) before randomization, but with the same patients as included in the study. The actual definition of the lower limit of 48 mm was deduced from a ROC analysis. The sensitivity of correctly classifying the patients with severe pain as well as the specificity of correctly classifying the patients with non-severe pain were both above 91 with this limit of 48 mm, as reported elsewhere. A particular strength of this analysis is that all calculations for the definition of ‘severe pain’ could be made independently of the data from the randomized study period and beforehand without knowing any results of the subgroup analyses using an objective decision procedure.
Like in the original study (7), women were over-represented compared to men in the subgroup. Based on the diagnostic criteria recorded most of the patients with severe headache attacks suffered from migraine. In a population-based study in Germany among patients with migraine, and in particular among the particularly severe migraine cases, women in Germany were affected three times more frequently than men. There are also significantly more women than men suffering from severe non-migrainous headache forms (5). In this respect, the subgroup with severe headache attacks can be seen as representative for the proportion of the general population that suffers from severe headaches.
In the overall study, 482 patients were treated with ASA + PAR + CAF and 128 patients were treated with placebo (7). Of these, 137 (28.4%) in the ASA + PAR + CAF group and 42 (32.8%) in the placebo group met the criteria defined for the subgroup analysis. In an independent study by Lipton et al. (16), the superior effectiveness of a fixed-dose combination with ASA (250 mg), paracetamol (250 mg), and caffeine (65 mg) versus placebo was already shown in patients with migraine. Patients met the criteria for severe, disabling migraine (post-hoc subgroup) if they reported a history of migraine attacks characterized by at least severe pain and severe disability, and treated attacks characterized by severe pain and at least severe disability (17). Of the original 1220 patients in the study, 172 (14.1%) fulfilled these criteria. In further studies with OTC analgesics, 40% of the ASA 1000 mg group and 37% of the placebo group (15), 45.0–50.5% of the ibuprofen (200–600 mg) groups (18), 28.8% of the paracetamol (1000 mg) group, and 29% of the placebo group (19) evaluated their headache at baseline, or the intensity of typical migraine pain without treatment as ‘severe’. Besides the expected variability, the higher proportion of patients with severe pain intensity can be explained by the fact that in order to belong to the current subgroup, all four criteria had to be fulfilled, which led to a reduction in the numbers. In this respect, the studies with OTC analgesics do not differ from those with triptans. For instance, in five different studies with triptans, 28–34% (20), 30–42% (21), 38% (22), 47–48% (23), and 37–47% (24) of patients described their headaches as ‘severe’ at baseline.
In this post-hoc analysis, the fixed-dose combination group was comparable to the placebo group regarding all baseline characteristics.
For all primary and secondary endpoints with the subgroup of severe headache pain and impairment, the fixed-dose combination ASA + PAR + CAF was statistically significantly superior to placebo. These findings confirm the analyses by Goldstein et al. (17), which show a statistically significant superiority over placebo both for the most severe segment of migraine attacks and for all endpoints (pain response at 2 h and 6 h after treatment, PID 0.5–6 h, and functional disability at 2 h and 6 h). No ‘conflicting results’ occurred either in our analysis or in the independent analysis of Goldstein et al. (17), and both the primary endpoints and all secondary endpoints point in the direction of a superior effectiveness of the fixed-dose combination with ASA, paracetamol, and caffeine, even though, as is to be expected in subgroup analyses, not all reached the statistical significance threshold. In terms of tolerability and adverse events, this subgroup roughly corresponds to the results of the general study (7), in which a side-effect profile comparable to another OTC analgesic was observable in the groups with active treatments (25).
For the complements too, the treatment with the fixed-dose combination was superior to a placebo treatment in all endpoints.
The strength of this post-hoc analysis is the operationalized definition of severe headache. A limitation is that these findings were based on a post-hoc subgroup analysis with multiple comparisons performed on a relatively small group of subjects. Reduced sample size and missing stratified randomization with the risk of low precision, and apparent heterogeneity in the estimates of treatment effects among subgroups as well as possible bias due to an imbalance with respect to prognostic factors are potential limitations for subgroup analyses in general. However, the results for this subgroup analysis are consistent with respect to all endpoints and to the patients with non-severe headache and the overall patient population.
In conclusion, the results of this subgroup analysis confirm that the fixed combination of ASA (250 mg), paracetamol (200 mg), and caffeine (50 mg) is effective and well tolerated in a broad spectrum from mild to severe migraine and tension-type headache severity independently of the headache diagnosis.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
HCD received honoraria for participation in clinical trials, contribution to advisory boards, or oral presentations from: Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, Coherex, CoLucid, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Grünenthal, Janssen-Cilag, Lilly, La Roche, 3 M Medica, Medtronic, Menerini, Minster, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brümmer, SanofiAventis, and Weber & Weber. Financial support for research projects was provided by Allergan, Almirall, AstraZeneca, Bayer, GSK, Janssen-Cilag, and Pfizer. Headache research at the Department of Neurology in Essen is supported by the German Research Council (DFG), the German Ministry of Education and Research (BMBF) and the European Union. HCD has no ownership interest and does not own stocks of any pharmaceutical company. HP and BA are employees of Boehringer Ingelheim Pharma, KG, Germany. The other authors have nothing to declare.