
Abstract
We investigated efficacy, safety, and tolerability of two tablets of the fixed combination of 250 mg acetylsalicylic acid (ASA) + 200 mg paracetamol + 50 mg caffeine (Thomapyrin®) in comparison with two tablets of 250 mg ASA + 200 mg paracetamol, two tablets of 500 mg ASA, two tablets of 500 mg paracetamol, two tablets of 50 mg caffeine, and placebo in patients who were used to treating their episodic tension-type headache or migraine attacks with non-prescription analgesics. For the primary endpoint ‘time to 50% pain relief’ in the intention-to-treat dataset (n = 1743 patients), the fixed combination of ASA, paracetamol and caffeine was statistically significantly superior to the combination without caffeine (P = 0.0181), the mono-substances ASA (P = 0.0398), paracetamol (P = 0.0016), caffeine (P < 0.0001) and placebo (P < 0.0001). All active treatments except caffeine differed significantly (P < 0.0001) from placebo. The superior efficacy of the triple combination could also be shown for all secondary endpoints such as time until reduction of pain intensity to 10 mm, weighted sum of pain intensity difference (%SPIDweighted), extent of impairment of daily activities, global assessment of efficacy. All treatments were well tolerated. The incidence of adverse events observed was low.
Introduction
Migraine with and without aura as well as tension-type headache, as defined by the International Headache Society (1), are very common diseases all over the world. The 1-year prevalence of migraine in adults is 6% among men and 15–18% among women (2). In surveys of the general population in North America and Western Europe, the 1-year prevalence of episodic tension-type headache ranged from about 30% to about 80% (3). The overwhelming majority of these patients used medication for their headache (e.g. 95% of men and 97% of women with migraine), but about two-thirds of migraineurs (4) and more than 80% of tension-type headache patients (5) never consulted a physician for their headaches and used over-the-counter (OTC) drugs for headache treatment (6).
For decades most headache patients used antipyretic OTC-analgesics, such as paracetamol (acetaminophen; APAP), acetylsalicylic acid (ASA), or non-steroidal anti-inflammatory agents, as well as fixed combination analgesics like the combination of ASA, paracetamol and caffeine to treat their migraine attacks or tension-type headache episodes (7–9). More than 90% of patients who used non-prescription fixed combination analgesics in Germany took it for headache treatment (10). When effective, OTC analgesics offer several advantages over prescription drugs, including easy access, lower cost, and fewer adverse effects (AEs) in comparison with prescription drugs (11).
In 1993, the Non-prescription Drug Advisory Board of the Food and Drug Administration recommended the classification of caffeine when combined with ASA plus paracetamol as a category 1 analgesic adjuvant –‘recognized as safe and effective’ (8). Caffeine shifts the dose–response curve to the left with an increase of analgesic potency of about 40% (8, 12). The observed synergism of ASA, paracetamol, and caffeine on the inhibition of PGE2 synthesis in microglial cells (13), a common model for the COX-2 inhibiting activity of non-steroidal anti-inflammatory drugs, may partly explain these effects. Caffeine alone might have analgesic properties for specific types of pain in humans (14–16) and in human experimental pain models (17), but the overall evidence from clinical studies is weak.
Clinical evidence supports the safety and efficacy of fixed combined analgesics with doses of 250–265 mg ASA, 200–265 mg paracetamol, and 50–65 mg caffeine per tablet. The optimal dose relationship of 1.0 : 0.8 : 0.2 (ASA : paracetamol : caffeine) was derived from pharmacological and clinical dose finding studies (12), with a recommended minimal dose of 50 mg caffeine per tablet. In patients with tension-type headache the three-fold combination analgesic was superior to both 1000 mg paracetamol and placebo (18). In patients with migraine with or without aura the combination was superior to placebo in the reduction of pain intensity, nausea, photophobia and phonophobia (19).
The key issue is whether the combination has a better therapeutic ratio (ratio of analgesia to side-effects) than either component, or produces a higher maximal analgesic efficacy at tolerable doses (20, 21). We therefore investigated the fixed combination of ASA, paracetamol, and caffeine in comparison with the combination without caffeine, with the monotherapies, and placebo in a randomized, placebo-controlled, double-blind study in patients representative of subjects who are used to treating their headaches with non-prescription analgesics. We purposely did not predefine headaches into migraine and tension-type headaches.
Methods
Patients
In this multicentre study, conducted between September 1998 and January 2003, male or female out-patients (18–65 years) were enrolled by practitioners and specialists in general and internal medicine throughout Germany. Patients who were not consulting for headache were asked whether they had headaches which they treated with non-prescription analgesics. Usual headaches had to meet International Headache Society (1) criteria for episodic tension-type headache (2.1) and/or migraine with or without aura (1.1, 1.2.1). They must have experienced these headaches for at least 12 months with a minimum of two headache episodes within the previous 3 months.
Patients meeting at least one of the following criteria were excluded: patient treats their headache with prescription analgesics or migraine drugs, patient requires higher single doses of non-prescription analgesics to treat their headache than indicated in the patient information leaflet, patient normally treats their headache with non-prescription analgesics in effervescent tablet form, headache occurs on more than 10 days per month or lasts untreated normally less than 4 h, close association between the occurrence of headache and menstruation (menstrual migraine). Further exclusion criteria were: concomitant treatment with prescription-only and/or non-prescription analgesics, antidepressants or antipsychotic medication (within the previous 4 weeks before study enrolment), antirheumatic or anti-inflammatory drugs that may influence the headache symptoms (within the previous 4 days), drugs containing ASA (above a daily dose of 100 mg/day), paracetamol or caffeine as well as migraine prophylaxis or administration of drugs that influence headache symptoms. Drug overuse connected with the headache (defined as the administration of analgesics or other drugs for the treatment of acute headache on more than 10 days per month) and/or alcohol or drug abuse as defined by DSM-IV were also exclusion criteria as well as pregnancy and lactation, gastrointestinal ulcers, pathologically increased bleeding tendency, glucose-6-phosphate dehydrogenase deficiency, hypersensitivity to paracetamol, caffeine, ASA, salicylates, and other anti-inflammatory/antirheumatic agents or other allergenic substances, bronchial asthma, concomitant treatment with anticoagulants, chronic or recurrent gastrointestinal symptoms, liver disorders, pre-existing renal damage, Gilbert's syndrome, or hyperthyroidism. A simultaneous participation and/or participation in another clinical trial within 4 weeks of entering this study was not allowed.
Prior to enrolment the patients gave their written informed consent according to sections 40, 41 of the German Drug Law (AMG) and ICH GCP (International Conference on Harmonisation, Guidance for Good Clinical Practice, E6) standards. Patients were allowed to terminate participation in the trial at any time, without giving reasons. The study was conducted in accordance with the Declaration of Helsinki, the German Drug Law (AMG), and ICH GCP standards and did not start before Independent Ethics Committee (IEC) approval was obtained. This trial complies with the Guideline for the clinical evaluation of analgesic drugs (22), with the Guidelines for controlled trials of drugs in migraine and with the Guidelines for trials of drug treatments in tension-type headache of the International Headache Society (23, 24).
Study design and treatments
This study was designed as a randomized, placebo-controlled, double-blind, multicentre parallel group trial. Three independent headache episodes were treated by every patient. Patients treated their first headache attack with their usual analgesic (open run-in phase) and recorded the features of the headache attack in a diary. The following two headache episodes were treated with the investigational study medication (treatment phase 1 and 2). Patients qualifying for this double-blind treatment phase were randomly allocated to one of the six treatment groups for both treatment phases (Table 1
Study treatments

Study medication was delivered by Boehringer Ingelheim Pharma GmbH & Co. KG, Germany. The doses corresponded to the maximal individual therapeutic single doses recommended for these analgesics. Blinding was ensured by using matched trial supplies, identical in colour, size, shape and taste. The trial medication was to be taken as a single dose when the headache occurred, and when the patients would normally have taken their usual analgesic.
Patients’ medical histories were recorded by the investigator at the screening visit. A structured questionnaire was used for the headache diagnosis. An independent headache expert (L.P.) performed a blinded classification of each of the three headache episodes for all study patients using this questionnaire.
Patients were allowed to use rescue medication 4 h after the administration of the trial medication if their pain remained; time of administration, the type of drug and the dose had to be documented by the patient in the patient diary.
Patients were instructed carefully to avoid any caffeine-containing beverages and/or food within 2 h before and within 4 h after administration of the medication. Otherwise, the type and quantity of such food or beverage had to be documented in the patient diary. Usual daily caffeine intake history was recorded at the screening visit.
Patients had to contact the investigator after each headache episode for their follow-up visit. The investigator reviewed the completed diary with the patient to ensure that all required information including safety and tolerability issues had been recorded.
Endpoints
The calculated time to 50% pain relief was chosen as primary endpoint based on the pain intensity recorded on a 100 mm visual analogue scale (VAS(PI)) (25). To this purpose, all patients recorded the pain intensity, prior to and then 30 min and 1, 2, 3 and 4 h after drug intake in the diary. The time to 50% pain relief was calculated by linear interpolation between adjacent observation time points. The first time point with 50% relief was used. At baseline the headache pain intensity had to be greater than 30 mm. Patients were asked to record date and time of administration in their patient diary to the nearest 5 min.
Secondary endpoints were:
Calculated time until reduction of pain intensity to 10 mm VAS(PI).
Percentage of patients with 50% pain relief at least after 2 h (evaluated on VAS(PI)).
Percentage of patients with 50% pain relief at least after 30 min, 1, 3 and 4 h (evaluated on VAS(PI)).
Pain intensity after 30 min, 1, 2, 3 and 4 h (evaluated on VAS(PI)).
%SPIDweighted [weighted Sum of Pain Intensity Difference (SPID) expressed as per cent of the maximum achievable SPID].
Extent of impairment of daily activities prior to and after 30 min, 1, 2, 3 and 4 h of drug administration (4-point verbal rating scale (VRS: ‘not impaired’, ‘somewhat impaired’, ‘greatly impaired’, ‘usual daily activities impossible’).
Global assessment of efficacy by the patient (4-point VRS: ‘very good’, ‘good’, ‘less good’, ‘poor’).
Global assessment of tolerability by the patient and investigator (4-point VRS: ‘very good’, ‘good’, ‘less good’, ‘poor’).
Safety assessment: recording of AEs [time of onset, duration and intensity of AEs; the intensity was determined by subjective evaluation of the patient and classified as mild (signs or symptoms easily tolerated), moderate (discomfort sufficient to cause interference with normal activities) and severe (incapacitating with inability to do work or undertake normal activity); the investigator determined the relationship between the drug treatment and AE].
Statistical analysis
The required sample size was estimated to be 1695 treated patients, giving the study a power of at least 0.80 to detect a difference between the triple combination and all other treatment groups assuming a difference in the average hourly success rate of approximately 20–40% (12) related to the primary endpoint. The smallest difference was expected for the comparison between the triple combination and the combination of ASA + PAR. The differences correspond roughly to a dose difference of half a tablet which is considered clinically relevant. The level of significance was set at 0.05 (two-sided). In order to take any premature withdrawals into account, the number of patients to be enrolled was set at 1870.
Data missing for any scheduled efficacy evaluation was replaced by the last observation carried forward procedure. If the baseline value was missing, the first measured value was used as baseline value. For patients requiring rescue medication, post-rescue medication pain intensity assessments were assigned the last recorded value before intake of rescue medication. The least favourable category (poor) was used for the global assessment of efficacy.
The safety endpoints were evaluated descriptively for each patient treated. The efficacy endpoints were evaluated comparatively for both an intention-to-treat (ITT, primary) and a per-protocol (PP, ancillary) dataset. The last headache episode was the basis of the evaluation of all efficacy endpoints. If a patient treated only the first headache with the study medication, this attack was analysed.
The Kaplan–Meier estimator was presented for the primary endpoint time to 50% pain relief and the log rank test was used to calculate P-values and test for statistically significant differences between the triple combination and all other treatment groups. In order to conclude superiority of the triple combination ASA + PAR + CAF to both the combination of ASA + PAR and all mono-substances and placebo, the P-values of all five pairwise comparisons had to be lower than the predefined significance level of 0.05 (two-sided).
The log rank test was also used to evaluate the time until reduction of pain intensity to 10 mm. An analysis of covariance model with factors centre, treatment group and the individual baseline value as covariate was used to evaluate the pain intensity difference (PID), the same model without covariate to evaluate the endpoint %SPIDweighted. The Cochran–Mantel–Haenszel test, stratified by the baseline extent of impairment, was applied to analyse the assessment of the extent of impairment of daily activities. Global assessment of efficacy and tolerability were made by means of the Wilcoxon rank sum test.
A review comprising data from approximately 600 patients was performed by an independent Data Monitoring Board as specified in the trial protocol. It was recommended to continue and analyse the study as described in this section. The evaluation of the quality and completeness of the data, identification of important protocol deviations and handling of problem cases were performed regularly and finally decided before locking and unblinding the database.
Results
Patient population
A total of 147 centres were initiated. Eight of them did not enter any subjects. Six centres submitted faked diaries and these data were disregarded. The remaining 133 centres enrolled 2336 patients (Fig. 1). Of these, 353 patients were withdrawn after the run-in phase evaluation mainly for loss to follow-up, violation of inclusion or exclusion criteria, consent withdrawn or inability to comply with the study procedures. Of the 1983 patients randomized, 94 did not take study medication. The remaining 1889 patients entered the investigational treatment phase and are part of the safety dataset. Finally, 146 patients did not return any diaries with VAS data or returned diaries with obviously unreliable entries, leaving a total of 1743 patients in the ITT dataset. The excluded patients came from centres who did not comply with GCP rules or where the diaries were filled in retrospectively or by another person. All these cases were confirmed by the investigator or study nurse and by independent quality assurance audits. The Ethics Committee was informed about all six centres who failed to comply with GCP rules. Patients with unresolved irregularities remained in the ITT dataset. However, the evaluation of the primary efficacy endpoint was repeated with all patients included (safety dataset) in order to assess the robustness of the primary analysis with the ITT dataset. Relevant protocol deviations in both treatment phases were recorded for 227 patients, leaving 1516 patients in the per protocol (PP) dataset. The most frequent protocol violations were use of prohibited previous or concomitant medication and caffeine intake outside the allowed time window.

Profile of subject disposition during the course of the study and inclusion in the analysis dataset. ∗Numbers do not add to total because some patients had several protocol violations.
Patient characteristics
Patient characteristics (Table 2) were comparable among all treatment groups. The over-representation of women (76% vs. 24% of men) reflects the overall higher proportion of women with migraine and episodic tension-type headache in the population. Based on the diagnostic criteria recorded, 84% of the patients usually suffered from migraine headache, 13% from episodic tension-type headache and 3% could not be classified. Without treatment, the usual pain intensity was severe or very severe in 62% and moderate in 37% of patients. The severity of pain was associated with inability to perform usual daily activities.
Patient characteristics and headache history (intention-to-treat dataset)

Patient characteristics of the treated headache were homogeneously distributed across all treatment groups (Table 3
Characteristics of treated headache (intention-to-treat dataset)

Efficacy results
For the primary endpoint ‘time to 50% pain relief’ with the ITT dataset (Fig. 2), the combination ASA + PAR + CAF was statistically significantly superior to the combination without caffeine (P = 0.0181), the mono-substances ASA (P = 0.0398), paracetamol (P= 0.0016), caffeine (P < 0.0001) and placebo (P < 0.0001). The median time to 50% pain relief was 1 h 5 min after treatment with ASA + PAR + CAF compared with 1 h 13 min, 1 h 19 min, 1 h 21 min, 1 h 47 min and 2 h 13 min after treatment with ASA + PAR, ASA, paracetamol, caffeine and placebo. All active treatments differed significantly (P < 0.0001) from placebo except caffeine (P = 0.4361). The additional evaluation with all treated patients included (safety dataset) confirmed the statistically significantly shorter time to 50% pain relief in patients taking the triple combination.
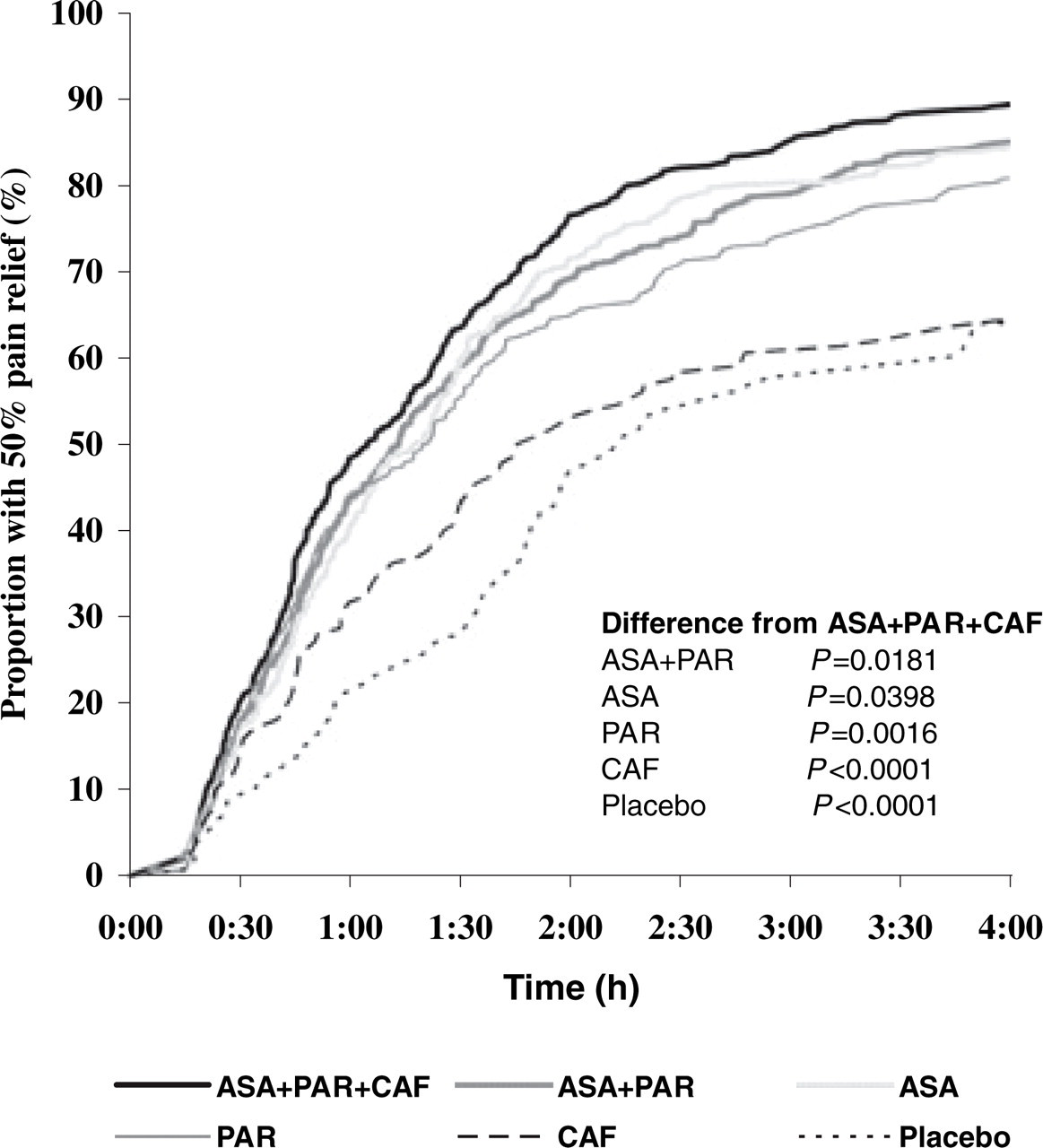
Primary efficacy endpoint – Kaplan–Meier estimator of the proportion of patients with 50% pain relief during the first 4 h after drug intake and P-values for differences from the triple combination (log rank test, intention-to-treat dataset).
Statistical analyses with the secondary endpoints corroborated the superiority of the combination ASA + PAR + CAF compared with both the combination without caffeine and all mono-substances and placebo. The time needed to reduce pain intensity to 10 mm was statistically significantly shorter in patients taking ASA + PAR + CAF compared with all other treatment groups. The median time was 1 h 56 min after treatment with ASA + PAR + CAF compared with 2 h 25 min, 2 h 31 min, 2 h 35 min, and 3 h 37 min after treatment with ASA + PAR, ASA, paracetamol, and caffeine. It lasted longer than 4 h after placebo. All active treatments were superior to placebo (P = 0.0002) except caffeine (P = 0.0936).
Pain intensity difference relative to baseline steadily increased over time in all treatment groups after intake of the medication (Fig. 3). This increase was more pronounced in patients treated with the combination ASA + PAR + CAF at all time points of pain intensity assessment after intake of the medication. A statistically significant difference was already seen after 1 h between the triple combination and all other medications except PAR. After 2 h, pain reduction in ASA + PAR + CAF was statistically significantly higher compared with all other treatments (Table 4). The improvement in pain intensity was very similar between the combination ASA + PAR and the mono-substances ASA and PAR within the first 2 h, separating slightly from 3 h onwards. The extent of pain reduction after intake of caffeine was clearly different from placebo, although it was at the same time clearly lower compared with the other active treatments. The resulting time interval weighted mean sum of pain intensity difference (%SPIDweighted) for ASA + PAR + CAF was significantly different from all other treatments except ASA.
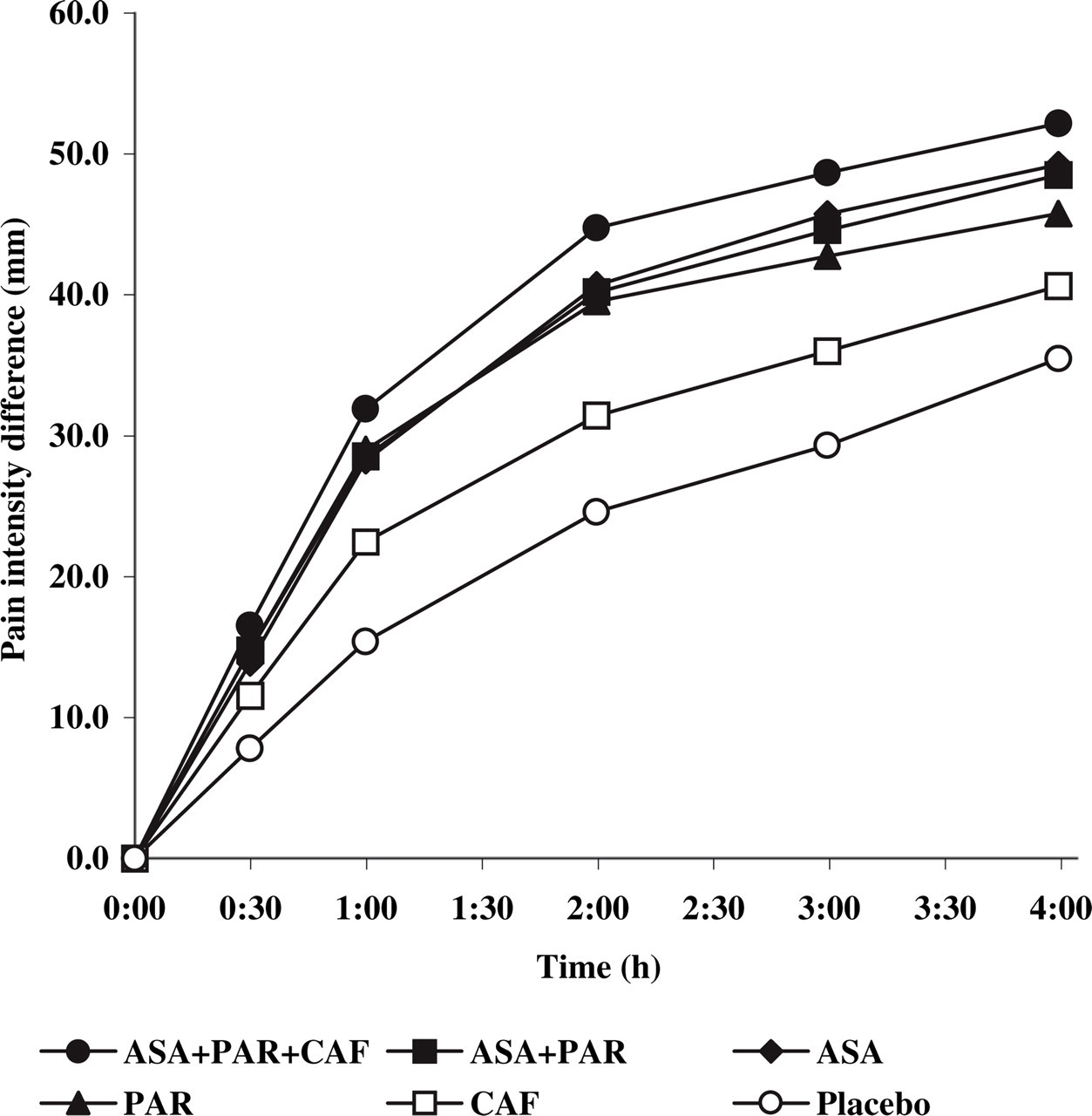
Time course of the mean pain intensity difference in the six treatment groups (intention-to-treat dataset).
Secondary efficacy endpoints (intention-to-treat dataset)

A larger proportion of patients treated with the triple combination did not experience relevant impairment of usual daily activities (Table 4). Differences from the triple combination were statistically significant compared with ASA, CAF and placebo.
The evaluation of global assessment of efficacy by the patients revealed a significant difference between the combination ASA + PAR + CAF and all other treatments.
Results of the efficacy endpoints with the PP dataset were similar to those for the primary analysis with the ITT dataset.
Safety and tolerability
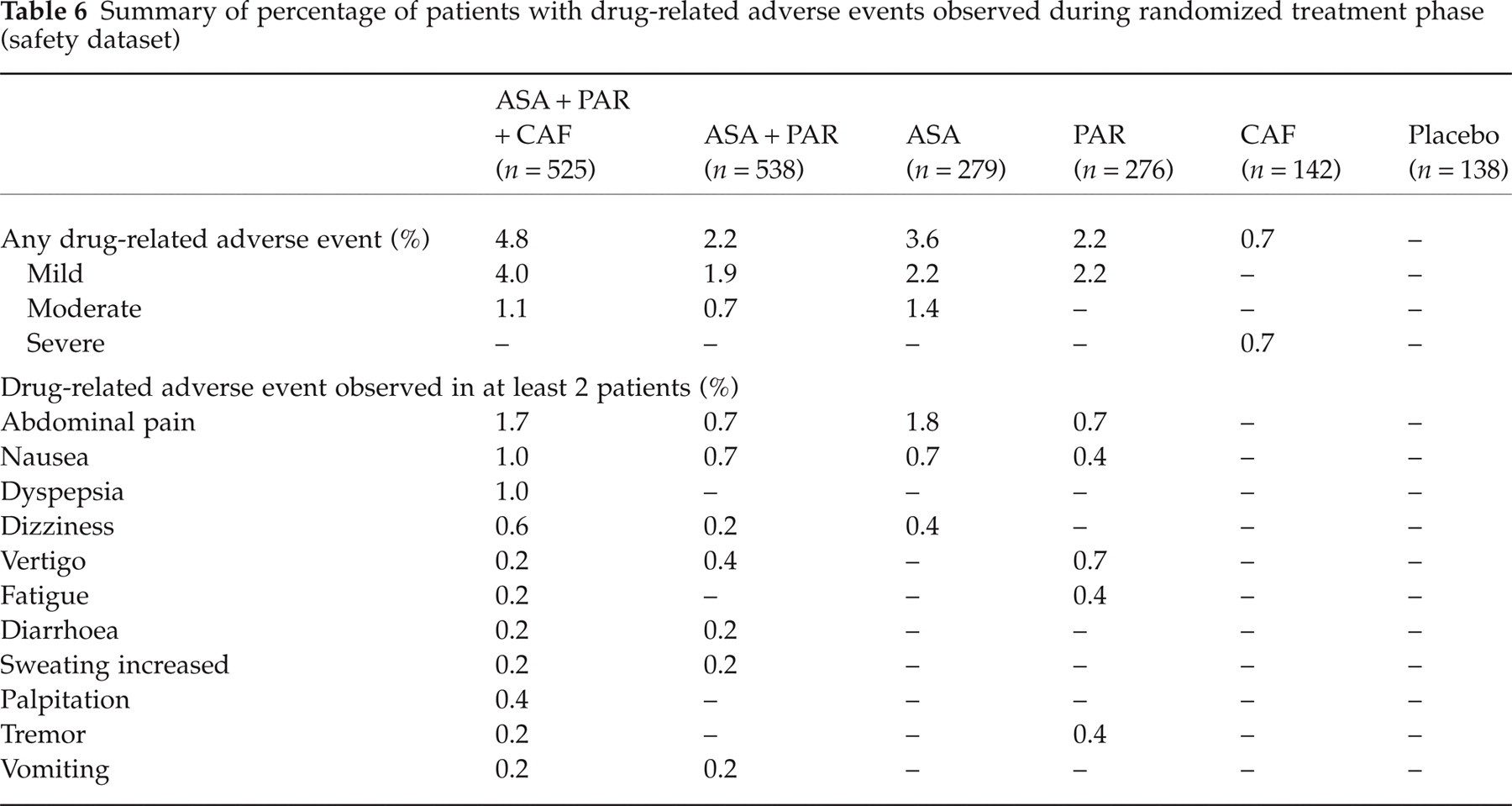
A total of 141 patients experienced AEs during the randomized treatment phase of the study (Table 5). The most frequent AEs reported were related to gastrointestinal system disorders, especially abdominal pain in all treatment groups (Table 5). The severity of all AEs was mild to moderate except in five cases, which were classified as severe (two ASA + PAR, one ASA, one PAR and one CAF). Fifty-two out of these 141 patients experienced AEs judged to be drug-related by the investigators (Table 6). Abdominal pain (reported by 20 patients), nausea (12 patients), dyspepsia, dizziness and vertigo (five patients each) were the most frequent drug-related AEs seen (Table 6). Two serious AEs were reported: one patient with acute enteritis of moderate intensity treated with ASA + PAR + CAF; one patient with an acute attack of ulcerative colitis of severe intensity treated with paracetamol. Both serious AEs were deemed by the investigator not to be drug-related.
Summary of percentage of patients with adverse events observed during randomized treatment phase (safety dataset∗)

Nine patients counted twice as they received two different investigational medications.
Summary of percentage of patients with drug-related adverse events observed during randomized treatment phase (safety dataset)
Global assessment of tolerability by the patients and investigators was very good or good in more than 90% of all patients.
Discussion
This large, double-blind, randomized, parallel-group, placebo-controlled trial with six treatment arms showed that the non-prescription fixed combination of ASA, paracetamol and caffeine is effective in treating migraine and episodic tension-type headache. Treatment differences for the primary and nearly all secondary endpoints consistently showed that the triple combination was significantly more effective than the combination of ASA plus paracetamol and more effective than either of these drugs given as monotherapy, caffeine or placebo.
The study was designed to include patients who would typically treat their headache with non-prescription drugs even if they did not know whether they suffered from migraine or tension-type headache. This reflects the typical situation of patients using non-prescription drugs for whom the diagnosis is unimportant but management of their headaches is vital.
Headache specialists do not see the kind of patients best suited for self-medication. Therefore, physicians in private practice were asked to recruit patients who consulted them for a reason other than headache. Not all study physicians were experienced in the conduct of clinical trials and this led to six centres being excluded due to major violations of GCP guidelines. If in individual cases compliance with the protocol was only moderate or poor, this caused an increase in the spread of the variables, reduced the chances of the detection of an actually existing treatment difference, and finally led to the underestimation of the superiority of the combination of ASA + PAR + CAF compared with the reference treatments.
We chose the calculated time to 50% pain relief measured by a visual analogue scale as the primary endpoint. It combines pain relief and time to its onset into a single endpoint, two aspects crucial for patients to assess the efficacy of the treatment. The sensitivity of the endpoint was confirmed, as all active treatments could be clearly separated from placebo with significant differences between placebo and all active treatments except for caffeine.
Time to 50% pain relief needs to be calculated for each headache episode separately. An originally planned procedure for multivariate testing of more than one endpoint (26) was proven to exceed the nominal significance level (27). Therefore, we decided to use the last headache attack for primary analysis in order to avoid a possible carry-over from the run-in phase treatment and to incorporate the final decision of the patient at the end of the two treatment phases. Irrespectively, the treatment with ASA + PAR + CAF resulted in highest and fastest pain relief in both treatment phases compared with the other study medications.
For the proof of the rationale of the fixed combination it would have been sufficient to use the dosages of the mono-substances as contained in the combination. We decided, however, to test the combination for superiority against dosages of the mono-analgesics proven effective in headache therapy and recommended for the treatment of migraine and of tension-type headache (9, 28–30). This decision implied that the study tested already for the superiority of analgesic potency, i.e. far more than the testing of the rationale of this combination. This explains the chosen dosage of 1000 mg ASA (factor 2.0 more than in the combination) and 1000 mg PAR (factor 2.5 more than in the combination). A previously probably underestimated aspect important to headache studies with antipyretic analgesics is the surprisingly flat dose–effect curve and a ‘plateau’ or ‘ceiling’ effect (31–34). This effect is independent of the chemical structure of the substance and applies to both the acidic and non-acidic antipyretic analgesics. In contrast, the synergistic effect of caffeine seen in this study when given in combination with ASA plus paracetamol confirms similar results from previous studies, where caffeine was added to ASA and/or paracetamol (18, 19, 35). It might be that the reported higher per capita consumption of analgesics in countries where no combination analgesics are available is due partly to this fact (36). The higher analgesic potency (more than efficacy) could possibly explain the clinical experience that the response rate of headache patients with the triple combination seems to be about three-quarters or above in comparison with about two thirds with mono-analgesics.
The study showed a moderate analgesic effect of 100 mg caffeine given as monotherapy compared with placebo. This had been observed in earlier studies (14–16). The results for the monotherapies were very similar to those reported in the literature (37).
The study included both patients with migraine and patients with episodic tension-type headache. Both conditions responded to therapy with analgesics (and caffeine). We adjudicated headache diagnosis by an independent neurologist but did not aim to diagnose aura symptoms. This study was deliberately not stratified and balanced with respect to the inclusion of patients with migraine and tension-type headaches according to the IHS criteria. Instead, the inclusion criterion was ‘headache treated successfully with non-prescription analgesics’. Because of the limited information, the unambiguous categorization of typical ‘OTC patients’ according to IHS criteria is difficult (38). Therefore, a procedure with a subsequent headache typing is particularly appropriate for headache studies with non-prescription analgesics if, in addition, each headache episode treated in the study is categorized according to the IHS criteria. There was a high consistency between the diagnoses of headache based on history and the actual treated headache episode.
Data on safety and tolerability, in particular those gathered in clinical trials, are important, especially concerning non-prescription analgesics. Safety is assessed on the basis of records of medically important side-effects, while tolerability refers to the extent of medically unimportant but clinically irritating side-effects of drugs (28), and it is crucial to distinguish between the two. The safety profiles of the fixed combination of ASA + PAR + CAF and of the reference treatments are very well known due to their world-wide million-fold use over decades. Overall it has to be considered as good or very good. All treatments were well tolerated and side-effects were less frequent than reported with ergotamine or triptans (39) and comparable to other OTC analgesics (40). Therefore, the triple combination has a favourable benefit–risk ratio. Cross-study comparisons of tolerability are difficult, because of their different recording methods (11, 31, 33, 37, 41). In some studies it remains unclear whether the data refer to any AEs or just to drug-related ones. Headache studies present the additional difficulty that it is often hard for both the patient and the investigator to differentiate between the very frequent concomitant symptoms of migraine and tension-type headache, such as nausea, vomiting and partly also abdominal pain, and drug-related AEs. This may to a certain degree explain the partly extremely great difference in the AE incidence reported in the literature. Overall, tolerability of these drugs can be considered as good; the true incidence of drug-related AEs may be a few per cent and only slightly higher than that for placebo.
A particular strength of the study was its large sample size. Nevertheless, an unbalanced randomization scheme was used for ethical reasons, with two to four times as many patients assigned to each group receiving active treatment as to groups receiving caffeine or placebo. Unbalanced randomization, however, favours placebo response (42) and may have worked against active drug. Including equal numbers of patients in the six treatment arms would have required a sample size of more than 3000 patients. Therefore, we decided on a design primarily powered for the test of the triple combination against the five other treatment regimens.
The results of this trial have important implications for clinical practice. Most prescription drugs for the treatment of migraine or tension-type headache are expensive and may have contraindications or relevant side-effects (9, 43, 44). Our study showed that the combination of ASA, paracetamol and caffeine (Thomapyrin®) can effectively and safely treat the pain and disability of episodic headaches and that this combination is significantly superior to monotherapy and the combination of ASA and paracetamol. Thus, the study verifies the evidence-based evaluations of the US Headache Consortium and of the DMKG (German Migraine and Headache Society), who in their most recent evidence-based therapeutic recommendation for the treatment of migraine (44) and for migraine and tension-type headache under self-medication (30) considered the fixed combination of acetylsalicylic acid, paracetamol and caffeine as the remedy of first choice.
Footnotes
Acknowledgements
We thank the participants in the study, and the doctors and medical staff in general practices throughout Germany who assisted with its conduct. We also thank the members of the Data Monitoring Board, Professor Dr G. Haag (Königsfeld) and Professor Dr L. A. J. Heinemann (Berlin), for their review of the study data as well as all members of the Boehringer Ingelheim Thomapyrin Study/CRA Team for their excellent work in conducting and data handling of this study. We thank Dr Anne MacGregor (London) and Professor Dr Richard Lipton (New York) for many valuable comments and suggestions. The study was sponsored by Boehringer Ingelheim Pharma GmbH & Co. KG, Vertriebslinie Thomae, Germany.