
Abstract
We assessed the efficacy and safety of oral single doses of 0.5 and 1 g metamizol vs. 1 g acetylsalicylic acid (ASA) in 417 patients with moderate episodic tension-type headache included in a randomized, double-blind, placebo- and active-controlled, parallel, multicentre trial. Eligibility criteria included 18–65 years of age, history of at least two episodes of tension-type headache per month in the 3 months prior to enrolment, and successful previous pain relief with a non-opioid analgesic. Treatment arms were metamizol 0.5 g (n = 102), metamizol 1 g (n = 108), ASA 1 g (n = 102) and placebo (n = 105). The analgesic efficacy of 0.5 and 1 g metamizol vs. placebo was highly statistically significant (α: 0.025; one-sided) for sum of pain intensity differences, maximum pain intensity difference, number of patients with at least 50% pain reduction, time to 50% pain reduction, maximum pain relief and total pain relief. A trend towards an earlier onset of a more profound pain relief of 0.5 and 1 g metamizol over 1 g ASA was noticed. All medications including placebo were almost equally safe and well tolerated.
Introduction
Headache is an extraordinarily frequent clinical symptom, which may be based upon a variety of illnesses or just be an expression of tension and fatigue. The International Headache Society (IHS) categorized headache disorders according to specific anamnestic and diagnostic criteria (1). Tension-type headache is the least characteristic of all headache types; its diagnosis is based mainly upon the exclusion of features characterizing other types of headache, such as the absence of one-sidedness, pulsatile pain, and worsening with physical activity. Moreover, tension-type headache may last for several days (2, 3). Common drug treatment of episodic tension-type headache is usually limited to simple analgesics or non-steroidal anti-inflammatory drugs (NSAIDs) that control most of the clinical symptoms. Non-drug treatment including physical activity and strategies for coping with stress are important in the management of this disease (4).
Metamizol (dipyrone) is a well-known non-opiate analgesic with antipyretic and spasmolytic qualities. The compound is readily hydrolysed in the upper gastrointestinal tract to methyl-aminoantipyrine (MAA). Subsequently, MAA is rapidly absorbed and transformed into various active metabolites, which are responsible for the pharmacological effects mentioned above. The analgesic action of metamizol reaches its maximum potency 40–60 min after ingestion, is effective for 6–8 h, which increases with the dose up to 1.5 g. Higher doses tend to prolong the analgesic effect (5). Metamizol has been proven to be clinically effective in the relief of pain due to acute renal colic (6), cancer (7), abdominal surgery (8) and dental extraction (9), and is comparably efficacious, if not superior, to NSAIDs and mild opioids. Systematic investigations with metamizol in episodic tension-type headache have not been performed until now. Thus, the present study investigated the efficacy, safety and tolerability of oral single doses of 0.5 g and 1 g metamizol and 1 g ASA relative to placebo in episodic tension-type headache.
Patients and methods
A total of 417 men and women with moderate episodic tension-type headache as defined by the IHS (1), participated in this randomized, double-blind, placebo- and active-controlled parallel group multicentre study. Each patient received either 0.5 g or 1 g metamizol, 1 g ASA or placebo. Medication was given orally as single doses. Two episodes of episodic tension-type headache, separated by an interval of at least 48 h, were to be tested. The study protocol was reviewed and approved by all relevant ethics committees in both Spain and Brazil. The entire study was conducted according to the principles of good clinical practice.
Patients aged between 18 and 65 years old were eligible if:
they experienced at least two episodes of tension-type headache per month in the 3 months prior to enrolment;
previous pain relief with a non-opioid analgesic was successful; and
the first episode of headache occurred before the age of 50 years.
Patients with relevant allergy or known hypersensitivity to the investigational drugs, its excipients and/or to other NSAIDs or with any other disorder contraindicating the administration of metamizol or ASA, i.e. bronchial asthma, peptic ulcer, gastroesophageal reflux, liver and/or renal dysfunction, were excluded. Other exclusion criteria were:
history of drug or alcohol abuse;
more than 15 episodes of tension-type headache per month;
tension-type headache that strongly correlated with the onset of hormone contraception;
prior (< 4 weeks) and/or concomitant treatment with antidepressant and/or antipsychotic drugs;
prior (less than 2 weeks) and/or concomitant treatment with NSAIDs, propranolol, metoprolol, flunarizine, cyclandelate, valproic acid, serotonergic antagonists, ergotamine, dihydroergotamine and benzodizepines;
use of benzodiazepines or any drug with analgesic properties in the 24 h prior to the intake of the study drug;
concomitant treatment with heparin or coumarin derivatives; and
ingestion of over-the-counter analgesics or any medication containing ASA and/or metamizol at the first signs of the tension-type headache episode under evaluation.
Pregnant women and nursing mothers were also excluded.
Treatment
Patients were randomly assigned to either 0.5 g metamizol or 1 g metamizol, 1 g ASA or placebo. Medication was given orally as single doses. The randomization was stratified by centres in blocks of four patients to ensure equally balanced distribution of the four treatment groups. The double-blind design of the study was achieved by using the double-dummy technique. The randomization scheme was generated using the software ClinPro LBL version 6.0 Clinical Systems Inc. The clinical trial supplies were packaged into a small box and comprised the randomized treatment for two episodes of tension-type headache. The individual patient received a single treatment at the beginning of the trial for the first episode and at visit 2 for the second episode of tension-type headache. All patients were instructed to take a single dose of study medication when headache was perceived as at least moderate. Subsequently, the patient had to record the headache pain intensity and pain relief over an observation period of 4 h in the diary provided. The decision on type and dose of rescue medication was at the discretion of the investigator and was allowed only 2 h after intake of the investigational treatment. The patients recorded the brand, dose and time of rescue medication in their diaries.
Measurements
The primary efficacy endpoint was defined as the time interval weighted sum of pain intensity difference (SPID) from baseline on a 0–100 mm visual analogue scale (VAS; 0 = no pain, 100 = unbearable pain). To this purpose, all patients recorded the pain intensity prior to and then 30 min and 1, 2, 3 and 4 h after drug intake in the provided diary. Ancillary derived endpoints included pain intensity difference (PID) at each time-point, maximum pain intensity difference (MAXPID) within 4 h after drug intake, the number of patients with at least a 50% pain reduction at all time-points and time to 50% pain intensity reduction. Simultaneously, all patients scored pain relief by means of a 5-point verbal rating scale (VRS; 0 = no relief; 1 = little relief; 2 = some relief; 3 = considerable relief; 4 = complete relief). These data were used to calculate the maximum pain relief (MAXPAR) within 4 h after drug intake and total pain relief (TOTPAR) up to 4 h after drug intake. Subsequently, investigator and patient judged the final global efficacy and the overall tolerability of the investigational treatment on a 4-point VRS (1 = good; 2 = satisfactory; 3 = not satisfactory; 4 = bad).
Incidence and nature of adverse events were recorded at each visit using non-specific and specific questions related to expected adverse events. Moreover, all patients were asked for time of onset, duration and intensity of the adverse event. The intensity was determined by subjective evaluation of the patient and classified as mild (signs or symptoms easily tolerated), moderate (discomfort sufficient to cause interference with normal activity) and severe (incapacitating with inability to do work or undertake normal activity). The investigator determined the relationship between the investigational treatment and adverse event.
Statistical analyses
Sample size calculation was based on the primary endpoint and results of similarly designed studies published by Migliardi et al. (10). A total of 400 evaluable patients (100 patients per group) were needed in order to have a power of 0.8 to detect a difference of 23% in the sum of pain intensity differences between treatments and assuming a loss to follow-up of 20%. The average over both headache episodes was the basis of the statistical evaluation of all endpoints except for the number of patients with at least a 50% pain reduction and the time to 50% pain intensity reduction which were analysed separately for each episode. Analysis of variance and t-tests were applied on sum of pain intensity difference (primary evaluation), pain intensity difference, maximum pain intensity difference and maximum pain relief within 4 h after drug administration and total pain relief over 4 h in a priori fixed ordering of hypothesis testing (superiority to placebo of metamizol 1 g first, followed by test of metamizol 0.5 g, and non-inferiority and superiority to ASA 1 g as a secondary objective). The Kaplan-Meier estimate and the log-rank test were used to analyse the time elapsed to 50% pain intensity reduction, and the Fisher's exact test and the Wilcoxon's test were used to compare the remaining secondary endpoints of the active treatment groups vs. placebo. The level of significance was set at P < 0.025 one-sided. Data are expressed as mean (
Results
Of 417 patients that were enrolled, 57 were randomized but did not take any study medication, the majority of them because no headache attack occurred during the study period. The study population consisted of 360 patients who were treated with either 0.5 g metamizol (n = 83) or 1 g metamizol (n = 95), 1 g ASA (n = 91) or placebo (n = 91). There were 31 participating centres; three centres included less than four patients. Four patients did not return their diaries and were therefore excluded from the efficacy analysis. The overall over-representation of women, 271 vs. 89 men, was judged to reflect the higher incidence of episodic tension-type headache in women. The other demographic characteristics, i.e. age, height, weight, body mass index, smoking history, alcohol consumption and coffee consumption, were comparable across all treatment groups.
The time-course of the mean pain intensity of both episodes of episodic tension-type headache is graphically displayed in Fig. 1. In total 326 patients treated both attacks. The patient's assessment of pain intensity at baseline was comparable for all treatment groups and decreased over time after administration. The pain intensity reduced steadily for all three active treatments in comparison with placebo up to 4 h after administration. Although the extent of pain reduction was more pronounced for 0.5 and 1 g metamizol, the treatment contrast vs. 1 g ASA gradually decreased beyond 1 h after administration. The resulting time interval weighted mean sum of pain intensity difference over both episodes reached 12.20, 12.64, 10.56 and 8.10 for 0.5 and 1 g metamizol, 1 g ASA and placebo, respectively. The statistical analysis by

Time course of the mean pain intensity in the four treatment groups according to the pain intensity assessments on the visual analogue scale. ○ Placebo; □ ASA 1.0 g; ▪ Metamizol 0.5 g; • Metamizol 1.0 g.
Ancillary statistical analyses of sum of pain intensity difference confirmed the non-inferiority of 0.5 g and 1 g metamizol to 1 g ASA (P < 0.0048) and a trend towards superior efficacy of 1 g metamizol over 1 g ASA (P = 0.0363) (Table 1). The mean maximum pain intensity difference from baseline was 4.11, 4.28 and 3.74 cm for 0.5 g and 1 g metamizol and 1 g ASA, respectively; the treatment contrast with placebo (3.04 cm) reached statistical significance for all active treatment groups (P < 0.0002 for metamizol groups and P < 0.0135 for ASA).
Analgesic efficacy of two doses of metamizol, acetylsalicylic acid (ASA) and placebo in patients with moderate episodic tension-type headache
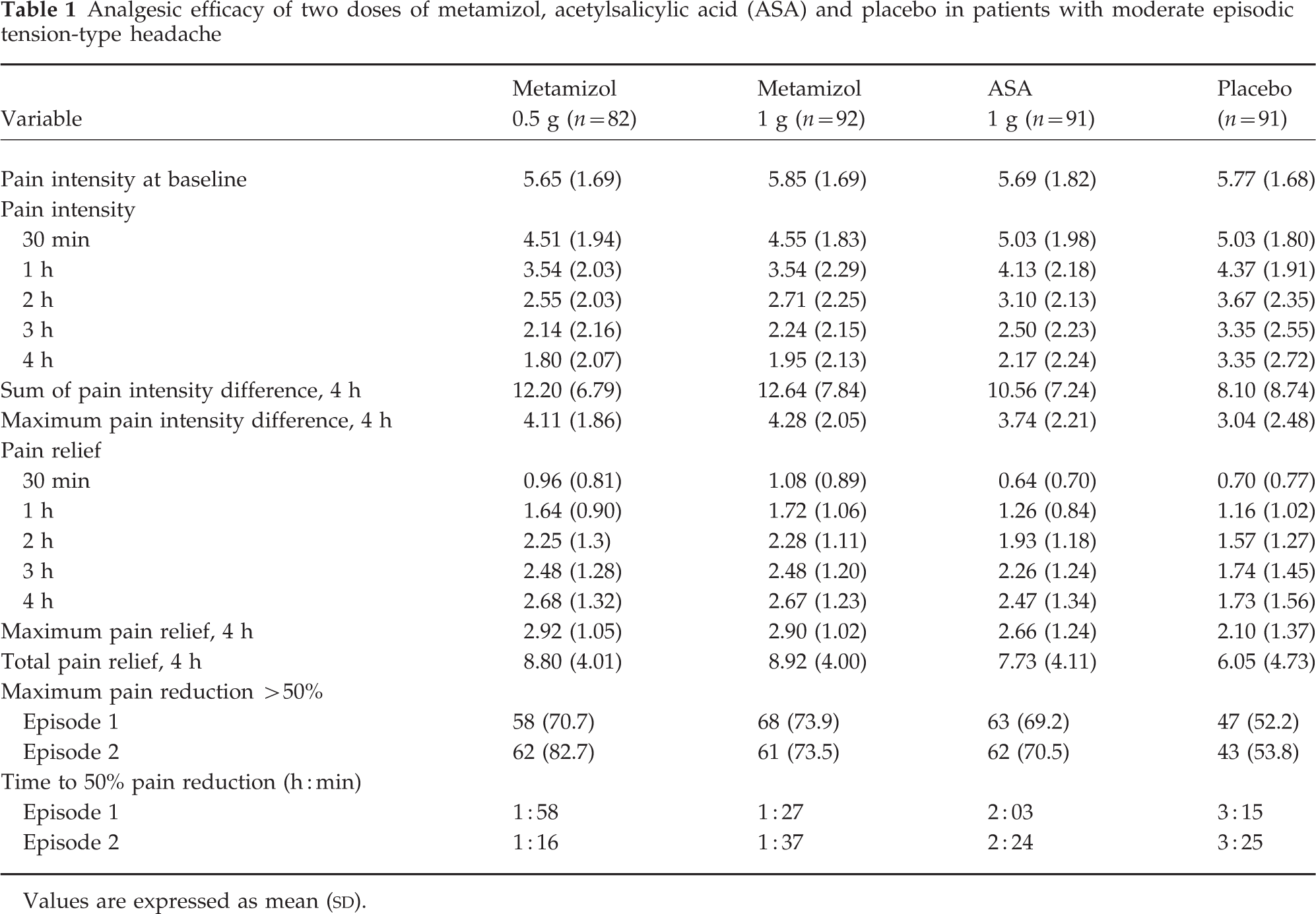
Values are expressed as mean (
The time-course of the mean pain relief of both episodes of episodic tension-type headache is graphically displayed in Fig. 2. Relative to placebo and 1 g ASA, initially a more pronounced pain relief under treatment with 0.5 and 1 g metamizol was observed up to 1 h after drug intake. Thereafter, pain relief improved steadily for all three active treatments in comparison to placebo up to 4 h after administration. Although the extent of pain relief remained superior for 0.5 g and 1 g metamizol along the entire observation period, treatment contrast vs. 1 g ASA gradually decreased beyond 1 h after administration. The mean maximum pain relief was 2.92, 2.90 and 2.66 for 0.5 and 1 g metamizol and 1 g ASA, respectively, and the corresponding differences to placebo (2.10) reached the level of statistical significance (P < 0.0001 for both metamizol groups and P < 0.0012 for ASA). The resulting mean total pain relief over 4 h was 8.80, 8.92 and 7.73 for 0.5 g and 1 g metamizol and 1 g ASA, respectively, with differences to placebo (6.05) statistically significant (P < 0.0001 for both metamizol groups and P = 0.0052 for ASA). The median time to a 50% pain reduction (h:min) in the first (and second) episode was 1:58 (1:16), 1:27 (1:37) and 2:03 (2:24) h:min after the administration of 0.5 g and 1 g metamizol and 1 g ASA, respectively, with differences to placebo (3:15 [3:25] h:min) that were statistically significant for both doses of metamizol in both episodes (P < 0.0145) and for 1 g ASA in the first episode only a trend was observed (P = 0.0350) (Table 1). The individual consistency for patients with a pain reduction > 50% in episode 1 and episode 2 was 88.9%, 82.0%, 82.0% and 81.0% for 0.5 g and 1 g metamizol, 1 g ASA and placebo, respectively.

Time course of the mean pain relief in the four treatment groups according to the pain relief assessments on the verbal rating scale. ○ Placebo; □ ASA 1.0 g; ▪ Metamizol 0.5 g; • Metamizol 1.0 g.
The percentage of patients per active treatment in the first (and second) episode using rescue medication was 15.9% (9.3%), 9.8% (14.5%) and 13.2% (14.8%) for 0.5 g and 1 g metamizol and 1 g ASA, respectively, which was lower than 27.8% (23.8%) observed under placebo. The percentage of patients per treatment in the first (and second) episode judging the treatment as good or satisfactory was 63.1% (78.6%), 77% (67%) and 57.2% (63.6%) for 0.5 g and 1 g metamizol and 1 g ASA, respectively, as compared with 47.2% (46.3%) for placebo. The comparison of the two metamizol groups with the ASA group was not significant. However, the corresponding differences to placebo in the first (and second) episode reached the level of P < 0.005 (P < 0.001) for both metamizol groups and P < 0.044 (P < 0.004) for ASA. The results of global assessment of efficacy by the investigator confirmed the patients' judgement of efficacy.
A total of 28 patients experienced adverse events during the treatment phase of the study (seven in the 0.5 g metamizol group, five in the 1 g metamizol group, eight in the 1 g ASA group, and eight in the placebo group). The severity of all adverse events was mild to moderate, and most of them (19 cases) involved the gastrointestinal tract. Twenty-two out of these 28 patients experienced adverse events judged to be drug-related (four in the 0.5 g metamizol group, five in the 1 g metamizol group, six in the 1 g ASA group, and seven in the placebo group). Dyspepsia, which was reported by 16 patients, was the most frequent drug-related adverse event seen. Six patients reported altogether: increased sweating (one treated with placebo); vomiting (one treated with placebo); somnolence (one treated with 0.5 g metamizol and one treated with placebo); nausea (one treated with 0.5 g metamizol, one with 1 g metamizol, one with 1 g ASA and one with placebo); urticaria (one treated with 0.5 g metamizol); and taste perversion (one treated with placebo). Global assessment of tolerability by the patients was good or satisfactory in more than 90% of all patients.
Discussion
Metamizol has widely been used for over 20 years in episodic tension-type headache, presumably under the umbrella of the well-known analgesic activity in other pain conditions. The present study confirms this empirical use in episodic tension-type headache as the data show metamizol to be an effective and safe analgesic in this indication. These findings are conditioned by the fact that eligibility criteria included successful pain relief with a non-opioid analgesic in previous episodes of tension-type headache. It is well established that NSAIDs are frequently used by patients to control most of the clinical symptoms, therefore it is not possible to extrapolate for patients resistant to NSAIDs. Even a single dose of 0.5 g metamizol was shown to be at least equipotent to the analgesic efficacy of 1 g ASA, which is today regarded as a well-established dose and known to be effective. In fact, it is the maximum allowed dose for this indication since in many countries 500 mg is being recommended as the standard treatment for this type of headache. A trend towards superior efficacy of 1 g metamizol over 1 g ASA was observed across most study parameters, including onset of action. This is also observed by the time to a 50% pain relief, which in comparison with 1 g ASA was considerably shorter under 1 g metamizol, i.e. 1:25 h:m vs. 2:03 h:m and 1:34 h:m vs. 2:26 h:m in episodes 1 and 2, respectively. These results are in line with previous observations, in which the early onset of analgesic action for metamizol was shown in pain models, such as third molar extraction and acute renal colic pain, i.e. 2 g metamizol produced a faster analgesic effect than did ibuprofen 600 mg and diclofenac 75 mg (6, 9).
The fast onset of action may perhaps be explained by the rate and extent with which the metabolites of metamizol cross the blood–brain barrier. Oral administration of 0.5 g metamizol led to a mean cerebrospinal fluid/plasma level ratio of 0.4 for the metabolite MAA only at 30 min after intake (11). Alternatively, unlike ASA and classical NSAIDs, metamizol slightly inhibits prostaglandin synthesis, suggesting that this drug may exert its analgesic properties via different pharmacological mechanisms. Two of such mechanisms are the following: (a) metamizol may desensitize the nociceptor activation in the peripheral nervous system; and (b) metamizol may have central anti-nociceptive properties by acting on the periaqueductal grey matter, thus activating inhibitory descending pathways to the spinal cord (12–15). Moreover, in vivo specific COX-2 inhibition has recently been suggested as an ancillary mechanism in order to explain the analgesic activity of metamizol (16).
The clinical efficacy of 0.5 g and 1 g metamizol during the 4-h observation period was comparable and therefore no dose relationship could be established. The very efficient pain resolution with the lower dose may account for the lack of difference between 0.5 g and 1 g metamizol. This finding is consistent with previous data in patients with colic pain given metamizol in whom 1 g metamizol reduced pain as effectively as 2 g metamizol during the first hour after administration and a minimum analgesic advantage in favour of the higher dose was not seen until 6 h after drug administration (6, 17). Whether 1 g metamizol offers a more sustained and profound pain resolution over 0.5 g metamizol in episodic tension-type headache beyond 4 h after administration can presumably not be addressed owing to the self-limiting nature of this pain phenomenon.
The nature, incidence and severity of adverse events reported under treatment with metamizol and ASA was well comparable to that of placebo treatment. Interestingly, the rate of gastrointestinal events with 1 g ASA was identical to that of placebo. The exclusion of patients with history of gastric intolerance to NSAIDs in this study may have accounted for this finding. In addition, the double-blind design of the trial required the double-dummy method, so that each patient was obliged to take a four-tablet dose per episode of tension-type headache, which may have caused the slightly higher incidence of some gastrointestinal discomfort in the placebo group. Differences with regard to the global assessment of tolerability by patients and investigators (including final assessment) were not observed.
It has been suggested that metamizol may induce blood dyscrasias, such as agranulocytosis. In the early 1970s the incidence of such blood dyscrasias due to metamizol was considered to be greater than 0.1%. However, no epidemiological studies on this issue had been available until publication in 1986 of the International Agranulocytosis and Aplastic Anemia Study (18), the results of which indicated that the excess risk of agranulocytosis was less than 1.1 per million users and that the risk of aplastic anaemia was virtually non-existent. Furthermore, metamizol is not related to upper gastrointestinal bleeding (19). A recent survey of population-based studies on the safety of metamizol and ASA showed a lower incidence of serious adverse drug reactions for metamizol (20). The benefit:risk ratio of metamizol has been discussed controversially; however, the more recent epidemiological view would suggest that metamizol has a total drug risk that is lower than that of aspirin.
In summary, treatment with 0.5 g and 1 g metamizol resolves pain due to episodic tension-type headache to a clinically relevant extent. A clear and consistent trend towards an earlier onset and more profound pain reduction and pain relief of either metamizol dose over 1 g ASA was observed. Metamizol and ASA were approximately equally safe and well tolerated.
Footnotes
Acknowledgements
The study was sponsored by Boehringer Ingelheim. The authors thank J.A. Pareja, J. Sánchez-Ojanguren, J.C. Martínez-Castrillo, J. Parra, O. González-Aznar, C. Ruiz-García, G. Friera, T. Manjón, A. Morales, A. López-Plana, L. Llosá, F. López-Expósito, R. Espona, S. Martínez, A. Bonillo, I. Alegre, X. Mundet, J. Casadevall, F. Luque, G. Baulíes, C. Rodrigo, M.L. Rodríguez-Morató, G. Amorós, E. Barraquer, G. Badell, E. Teixidó, P. Esteras, D. Lumbreras, A. Rivera, A. Cama, M. Cobos, N. Martínez-León, M.J. Font, I. González-Saavedra, A. Luque, M. De la Figuera, M.J. Queijas, C. Bordini, J.M.F. Almeida, L. Fuertes, C. Bustamante and R.M. Martínez. We thank Marta Pulido MD for editing the manuscript and editorial assistance.