
Abstract
Objective: To study the link between nonverbal learning disorder and right cerebral hemisphere dysfunction due to migraine attack in a case of Familial Hemiplegic Migraine.
Background: Familial Hemiplegic Migraine can cause neuropsychological deficits besides the motor ones. The nonverbal learning disorder is thought to be caused by a right hemisphere dysfunction.
Methods: We describe a child with Familial Hemiplegic Migraine type 2 who showed a transient neuropsychological impairment featuring a nonverbal learning disorder during and after a Hemiplegic migraine attack.
Results: Clinical and neuropsychological data showed a nonverbal learning disorder. A mutation in the ATP1A2 gene on chromosome 1q23 was found. Symptoms of nonverbal learning disorder outlasted the left hemiparesis. Two months later he showed a full recovery. Neurophysiological and neuroradiological evaluations were congruent with clinical course and with right hemisphere involvement.
Conclusion: The link between nonverbal learning disorder and right cerebral hemisphere dysfunction due to migraine attack is confirmed. Familial Hemiplegic Migraine can cause transient complex neuropsychological syndromes that can be overlooked if not appropriately investigated.
Introduction
Familial Hemiplegic Migraine (FHM) is a rare, genetically heterogeneous autosomal dominant disorder, characterized by migraine attacks with aura including a gradual progression of hemiparesis or other neurological deficits, which can outlast the headache (1). Mutations in three genes, including brain-expressed ion channels and a pump protein, have been identified in families with FHM. Mutations in the CACNA1A gene at chromosome 19p13 (FHM1) are responsible for 50–70% of FHM families (2). The other two genes involved are ATP1A2 at 1q23 (FHM2) and SCN1A at 2q24 (FHM3) (3,4). Clinically, the three types of FHM are similar except for cerebellar symptoms, which are usually present only in FHM1 (5). Epileptic seizures, although observed as part of the phenotypic spectrum in all the three forms, are more frequent in FHM2 (6,7). Neuropsychological impairment can be part of the neurological deficit during a migraine attack but its accurate assessment is difficult because of its quick recovery and because it can be obscured by major neurological deficits.
We describe a child with transient neuropsychological impairment resembling a nonverbal learning disorder or nonverbal learning disability (NLD) during and after a hemiplegic migraine attack. NLD is a developmental neuropsychological entity that was first described by Johnson and Myklebust in 1967 (8). Its current definition is mostly due to the research work made by Rourke several years later (9,10). Children suffering from NLD show a number of specific, potentially debilitating symptoms. Rourke has grouped these into three major areas: neuropsychological deficits, academic deficits, and social-emotional/adaptational deficits (10). Neuropsychological deficits include difficulties with tactile and visual perception, psychomotor coordination, tactile and visual attention, nonverbal memory, reasoning, executive functions, and specific aspects of speech and language. As a consequence of these deficits a discrepancy between Verbal-Performance IQ scores, with higher Verbal IQ than Performance IQ, is often present. Although this discrepancy alone cannot be diagnostic in the absence of other supporting evidence, it is common to find a discrepancy of at least 15 points between Verbal and Performance IQ scores and it is not unusual to find differences up to 40 points or more. Academic concerns regard deficits in math calculations, reading comprehension, specific aspects of written language, and handwriting. Social deficits include problems with social perception and social interaction (10).
Case report
A 9-year-old, right-handed boy was admitted at hospital with left facio-brachio-crural paresis, confusion and headache. The event started 24 hours earlier with intense headache, nausea and emesis, followed by paraesthesia of the left arm progressing to hemiparesis. He had had a similar, but shorter, event at 6 years of age, which had lasted 2 hours. A CT scan performed immediately after that attack was normal. An MRI performed 2 days later was normal. Familial history was positive for migraine with hemiparesis (three generations in the maternal line with vertical transmission). Besides the migraine, the patient and his family members had no other relevant medical history. The family members had never had either permanent or transient neuropsychological deficits.
The neurological examination at the time of admission revealed left hemiparesis. He was alert and oriented, answering correctly to questions in a monotonous voice; he avoided direct gaze and he showed an emotionless facial expression. He showed agnosognosia, as he spoke a lot about trivial matters without being worried about his hemiparesis. Headache was no longer present. Routine laboratory investigations from blood and urine samples were within normal limits. A brain CT scan showed oedema in the right cerebral hemisphere. An EEG showed significant right hemispheric slowing with no epileptiform activity. A brain 1.5 Tesla MRI, performed on the same day, confirmed the swelling of the right hemisphere due to cytotoxic oedema but no sign of ischemic stroke.
In the following 2 days in hospital he had two more episodes of headache without worsening of the neurological signs (hemiparesis was slowly recovering). A non-steroidal anti-inflammatory agent was administered (acetylsalicylic acid) with recovery of headache.
On day 4, he had a brief generalized seizure, which started with bilateral arm paraesthesia followed by consciousness impairment, ocular fixity and mild strength deficit in right arm. The EEG, performed a few minutes after the seizure, also showed electrical slowing in the left cerebral hemisphere, although less significant that in right one. Sodium valproate was started.
On day 5 MRI was repeated and it showed a worsening of right hemispheric swelling with mild deviation of septum pellucidum and compression of right lateral ventricle and third ventricle (Figure 1). MR spectroscopy (A) First MRI examination (day 1): top row, diffusion weighted imaging (DWI); bottom row, apparent diffusion coefficient (ADC) map. Cytotoxic oedema can be seen in the right peritrigonal white matter, corresponding to DWI hyperintensity and ADC hypointensity. Right hemisphere swelling can be seen, with evidence of reduced sulcal volume. (B) Second MRI examination (day 5): top row, DWI; bottom row, T2-fluid-attenuated inversion recovery (T2-FLAIR). There is reduction of the cytotoxic oedema and increased right hemisphere swelling with more pronounced sulcal asymmetry. (C) Third MRI examination (day 60): top row, DWI; bottom row, T2-FLAIR. There is complete recovery from cytotoxic oedema and brain swelling without evidence of gliosis.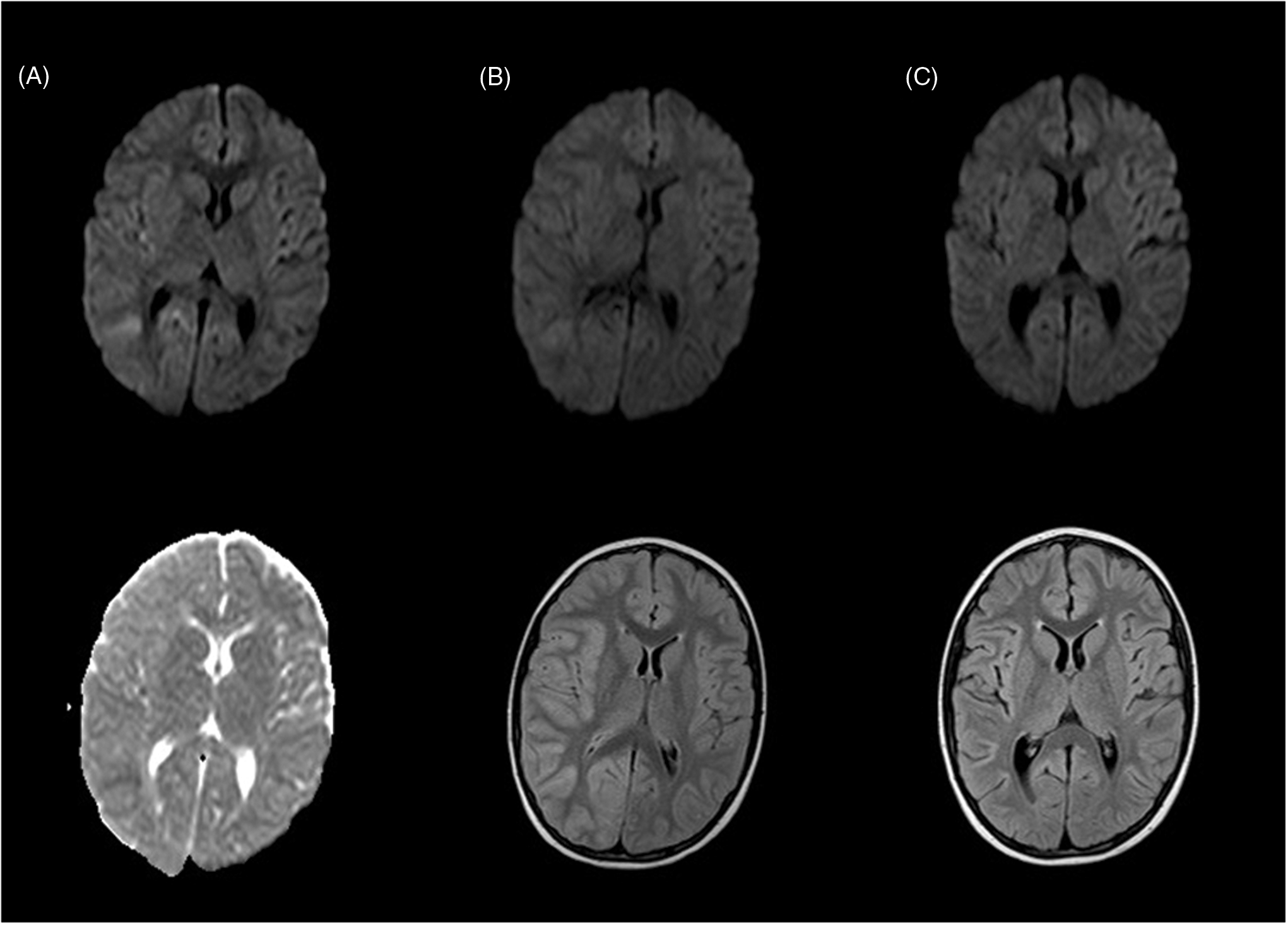
Sequencing of CACNA1A did not detect any mutation. Genetic analysis by direct sequencing of exons of encoding region of gene ATP1A2 detected a heterozygotic mutation, c.1816 G > A (pAla606Thr).
Set of tests used for neuropsychological assessment

WISC-III test (performance items)

Test of visual perception and visual-motor integration (TVP)

Tests of digital gnosias and right-left orientation

Memory and attention tasks

Academic and social difficulties were no longer present; especially notable was the improvement in mathematics. His behaviour was completely normal. His speech was completely adequate and his facial expression showed normal emotional changes. He was continuing to take sodium valproate as migraine prophylaxis. No headache or seizure had occurred in the past 24 months. MRI and MR spectroscopy, performed 10 months after the episode, were completely normal.
Discussion
Our young patient suffers from FHM type 2 due to a mutation in the ATP1A2 gene. Unfortunately, we could not perform a genetic analysis on the rest of the family, because we had no research funds to do so. Mutations in the CACNA1A and SCN1A genes can also cause FHM. Although different genes are responsible, the three forms have similar clinical symptoms: migraine attacks with aura and neurological deficits. In type 2 FHM epileptic seizures are relatively common (6,7), whereas in type 1 cerebellar dysfunction is often present (5).
FHM2 families present a wide phenotype besides theoccurrence of epileptic seizures. Epilepsy itself, described since the mapping of FHM2 gene (11), is not specific: generalized epilepsy such as absence epilepsy and generalized tonic-clonic seizures (12–14), focal epilepsy (7,15), febrile seizures (16,17) and benign familial infantile convulsions (6) have been reported. Recurrent transient episodes of coma or impaired consciousness have been described in FHM2 (15,18,19). Permanent neurological abnormalities can also be present in the FHM2 phenotype: mental retardation (19,20) and unusual motor disorders such as extra-pyramidal rigidity of the limbs and tongue apraxia (21) have been reported. An association with alternating hemiplegia of childhood has been argued (22,23).
Although transient neuropsychological impairment is not uncommon during a migraine attack (24), we found only one report describing brief unilateral spatial neglect during a hemiplegic migraine attack in a child revealed by administration of a neglect test and observation of drawing (25). Data collected from clinical examination, neuropsychological testing and teachers’ reports showed that our patient had a transient condition similar to NLD. Symptoms featuring NLD outlasted hemiparesis and are congruent with the current hypothesis about the involvement of the right hemisphere in the origin of NLD. According to Rourke (10), the NLD syndrome would be expected to develop under any set of circumstances that interferes significantly with the functioning of right hemisphere systems.
A MRI performed on day 5 showed oedema of the right hemisphere and the spectroscopy revealed reduction of peak of NAA in the white matter of the right parietal region. Magnetic resonance angiography and transcranial Doppler ultrasound were not performed. We know that in FHM symptoms are probably associated with neuronal metabolic dysfunction (26). The observed NAA reduction supports this hypothesis. NAA is found exclusively in neurons, and a decrease in this metabolite is widely considered to reflect neuronal loss or dysfunction (27). The latter is likely in our case as the NAA peak was normal and symptoms were no longer present 10 months later.
The ATP1A2 gene mutation affects the Na+/K+ ATPase pump, leading to a loss of function of the Na+/K+ pump (3). A reduction in the exchange of intracellular Na+ for extracellular K+ can depolarize neurons and make them hyperexcitable. Any further increase in extracellular K+ may facilitate cortical spreading depression, the probable mechanism for aura in FHM, or may start an epileptic seizure. A mutation in the same gene was responsible for the presence of both FHM and benign familial infantile convulsion in a family reported by Vanmolkot et al. (6).
Our case shows that, in addition to hemiparesis and other neurological deficits, FHM can cause transient complex neuropsychological impairments that can be overlooked if not appropriately and precociously investigated. Early and continued neuropsychological testing could bring new findings and contribute to a better definition of this disorder. Furthermore, our case, a kind of in vivo model, supports the role of right hemisphere in producing the constellation of NLD symptoms.
Footnotes
Acknowledgements
We are grateful to Craig Mathis for his review of the English version.
Conflicts of interest
The authors declare that there is no conflict of interest.
Patient consent
Parents’ written approval of publication was obtained.