
Abstract
Background: The development of new agents for the preventive treatment of migraine is the greatest unmet need in the therapeutics of primary headaches. Topiramate, an anticonvulsant drug, is an effective anti-migraine preventive whose mechanism of action is not fully elucidated. Since glutamate plays a major role in migraine pathophysiology, the potential action of topiramate through glutamatergic mechanisms is of considerable interest.
Methods: Recordings of neurons in the trigeminocervical complex (TCC) and the ventroposteromedial thalamic nucleus (VPM) of anesthetized rats were made using electrophysiological techniques. The effects of intravenous or microiontophorezed topiramate on trigeminovascular activation of second- and third-order neurons in the trigeminothalamic pathway were characterized. The potential interactions of topiramate with the ionotropic glutamate receptors were studied using microiontophoresis.
Results: Both intravenous and microiontophorized topiramate significantly inhibited trigeminovascular activity in the TCC and VPM. In both nuclei microiontophoretic application of topiramate significantly attenuated kainate receptor-evoked firing but had no effect on N-methyl-
Conclusion: The data demonstrate for the first time that topiramate modulates trigeminovascular transmission within the trigeminothalamic pathway with the kainate receptor being a potential target. Understanding the mechanism of action of topiramate may help in the design of new medications for migraine prevention, with the data pointing to glutamate-kainate receptors as a fruitful target to pursue.
Introduction
Migraine is a disabling (1), costly (2) brain disorder (3) affecting about 12–15% of the population (4). The development of the triptans, serotonin 5-HT1B/1D receptor agonists (5), in the late 1980s provided a revolution in acute therapy with the availability of several very effective treatments (6). Preventive therapy remains somewhat underdeveloped, with an estimate that only one in three patients suitable for prevention receive such therapy in the USA (7). With the exception of methysergide, which is certainly efficacious (8,9) although with very considerable side effects (10), as yet no drug has been successfully developed that was specifically designed for migraine prevention. While there are choices for preventive therapy, such as beta-blockers, serotonin antagonists, calcium-channel blockers, anticonvulsants, or botulinum toxin (11), all have side effects or limitations that restrict their utility in many patients. Among these choices, topiramate, a derivative of the naturally occurring monosaccharide
The efficacy of topiramate in migraine prevention is well established (16). In multicenter randomized, double-blinded, placebo-controlled trials, topiramate reduced migraine periods in episodic (17–19) and chronic migraine (20,21). In patients who have migraine with aura it reduces aura frequency in parallel with headache reduction (22). Topiramate may have some persistence of effect after discontinuation (23), although it did not prevent progression from high-frequency episodic migraine to chronic migraine (24).
A number of possible mechanisms of action have been suggested to explain topiramate's anticonvulsant properties. These include modulation of the action of GABA on GABAA receptors (25,26), a negative modulatory effect on L-type high-voltage-activated Ca2+ channels (27), blockade of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate glutamate receptors (28,29), inhibition of the II and IV carbonic anhydrase isozymes (30), and a less prominent voltage-sensitive, use-dependent Na+ channel blockade (31). However, which of these pharmacological properties of topiramate contribute to its anti-migraine activity has not yet been determined.
Of the proposed anticonvulsant mechanisms of action of topiramate, its effects on glutamate receptors are of interest given current understanding of migraine pathophysiology. For both migraine with and without aura, a recent multicenter genome-wide association study identified the involvement of locus 8q22.1, which is located between two genes implicated in glutamate homeostasis in the peripheral circulation and in the brain (32,33). These genes are the plasma glutamate carboxypeptidase gene (PGCP) and the astrocyte elevated gene 1 (MTDH). PGCP encodes for the plasma circulating glutamate carboxypeptidase which has an important role in the hydrolysis of circulating peptides to glutamate, while MTDH down-regulates the SLC1A2 gene which is responsible for encoding the glutamate transporter-1 (GLT-1; EAAT2 in human), the main astroglia-specific glutamate transporter. Glutamate drives excitatory neurotransmission in key brain areas involved in migraine pathophysiology and its levels are increased during a migraine attack both at the periphery and in the CNS (34). As glutamate plays a major role in migraine pathophysiology, modulation of glutamate receptors may have significant therapeutic potential (35).
In the current study we aimed to evaluate the effects of topiramate on trigeminovascular activation within the trigeminocervical complex (TCC) and the ventroposteriomedial thalamic nucleus (VPM), which are the principal sensory nuclei where transmission of trigeminovascular nociception is relayed within the ascending pathway (36). We further aimed to characterize the actions of topiramate on the ionotropic glutamate receptors N-methyl-
Materials and methods
General procedure and surgery
All studies were conducted and terminated under general anesthesia in accordance with guidelines of the University of California, San Francisco, Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (250–400 g; n = 43) were anaesthetized with pentobarbitone sodium (60 mg/kg i.p.) and prepared for physiological monitoring. The right femoral vein and artery were cannulated for administration of supplementary anaesthesia (20–25 mg/kg pentobarbitone sodium infusion) and monitoring of blood pressure, respectively. The left femoral vein was further cannulated for intravenous administration of topiramate or its vehicle control. The trachea was intubated for artificial ventilation with oxygen-enriched medical air (Ugo Basile, Coerio, VA, Italy), before the head of the animal was fixed in a stereotactic frame (Kopf Instruments, Tujunga, CA, USA). Core temperature was monitored and maintained using a rectal thermistor probe linked to a homeothermic heater blanket system (TC-1000, CWE Inc., Ardmore, PA, USA). A sufficient depth of anesthesia was judged from the absence of withdrawal reflexes, and gross fluctuations in blood pressure measurements. End-tidal CO2 was continuously monitored and maintained between 3.5 and 4.5%. All physiological measurements were displayed on an online data analysis system (CED spike2v5 software; CED, Cambridge, UK). At the end of each experiment, animals were given a lethal dose of pentobarbital sodium (Fort Dodge Animal Health, Southampton, UK).
For recordings within the TCC, the skull was exposed and a craniotomy of the left or right parietal bone was performed with a saline-cooled drill to provide access to the dura/middle meningeal artery (MMA), which was then covered in mineral oil. A hemi-C1 laminectomy was carried out ipsilateral to the exposed MMA and the dura mater was incised to expose the brainstem at the level of the caudal medulla. For recordings within the VPM a small craniotomy was performed to expose the superior sagittal sinus (SSS) and the right or left parietal region above the VPM. The dura mater was incised and reflected to expose the cortex before the area was covered in mineral oil to prevent desiccation of the cortex and provide electrical insulation.
Stimulation of dural vessels and recordings from the trigeminocervical complex and the ventroposteromedial thalamic nucleus
Bipolar platinum stimulating electrodes were placed onto the MMA or the SSS and square-wave pulses with the lowest possible stimulus intensity were used to activate trigeminal nociceptive afferents (50–150 µA; 0.5 Hz; 100–200 µs duration; Grass Instruments S88 Stimulator, West Warwick, RI, USA). Extracellular recordings were made from second-order neurons in the region of the TCC or third-order neurons within the VPM, using microiontophoretic electrodes (Carbostar 7S, Kation Scientific, Minneapolis, MN, USA) consisting of a seven-barrelled glass pipette and incorporating a carbon-fiber recording electrode (impedance 1 kHz: 0.4⊟0.8 MΩ). Extracellular recordings from neurons in the VPM were made according to the stereotactic coordinates derived from the brain atlas of Paxinos and Watson (38). The electrode was slowly lowered into the corresponding nucleus at 5 µm increments with a hydraulic microstepper (David Kopf Instruments, Tujunga, CA). The signal from the recording electrode was fed via a headstage amplifier (NL100AK; Neurolog, Digitimer, Herts, UK), AC preamplifier (Neurolog NL104A, gain x1000) and noise eliminator (Humbug, Quest Scientific, North Vancouver, BC, Canada) to Neurolog filters (NL125; bandwidth typically 700⊟10 kHz) and thence to a second stage variable amplifier (Neurolog NL106, gain typically × 50⊟90). This signal was fed to a gated amplitude discriminator (Neurolog NL201). The filtered and amplified signal was displayed on an oscilloscope (OS 7020A, Goldstar Precision Co., Korea) and also fed to an audio amplifier (Neurolog NL120) to assist with the discrimination of single unit activity from background noise. A personal computer running Spike 2 software (Cambridge Electronic Design, Cambridge, UK) was used to collect and analyse electrophysiological data. Post-stimulation histograms were constructed on line and saved to disc, in order to record the response of units to electrical stimulation of the dura/vessel complex. The sum of the data for a total of 25 stimuli were displayed in post-stimulus histograms, while background cellular activity was continuously monitored via peri-stimulus histograms. Neuronal action potential firing in response to microiontophoresis of glutamate receptor agonists was analyzed as cumulative rate histograms.
Receptive field characterization
Receptive fields were sought on the ipsilateral craniofacial region for all cells recorded in the TCC and on the contralateral craniofacial region for cells recorded in the VPM. The cutaneous receptive field was assessed in all three divisions of the trigeminal innervation and identified as the electrode was lowered into the area of interest. The receptive field was characterized for non-noxious (gentle brushing with a blunt probe) and noxious (pinch with toothed forceps) responses. Cells were classified as low threshold mechanoreceptors if they responded only to non-noxious stimuli, nociceptive specific (NS) if they responded to noxious stimulation only and wide dynamic range (WDR) if they responded to both.
Drugs
Topiramate hydrochloride was a kind gift from Johnson & Johnson PRD (Raritan, NJ, USA). Freshly prepared solutions of topiramate dissolved in distilled water for injection was intravenously infused 5 minutes after three consistent baseline responses of second, or third-order neurons to electrical stimulation of the dural vessels were obtained.
Seven-barrel carbon-fiber electrodes were used to deliver microiontophoretically freshly prepared solutions of glutamate receptor agents via a microiontophoresis current generator (Dagan 6400, Dagan Corporation, MN, USA). Micropipette barrels were filled with 50 mM of topiramate hydrochloride, 25 mM of the GluK1 (former GluR5; 39) kainate agonist iodowillardiine (Tocris), 25 mM of the AMPA agonist fluorowillardiine (Tocris), 100 mM NMDA (Tocris) and with pontamine sky blue dye (Gurr 6BX, BDH Laboratory Supplies, Poole, UK; 2.5% w/v in 100 mM sodium acetate). All compounds were ejected as anions (5⊟100 nA) and retained with small positive currents. Control represents the ejection of OH− ions, since the pH of dH2O was adjusted by the addition of 0.01 M NaOH. Current balance was provided through a barrel containing 200 mM NaCl. Microiontophoretic barrels had resistances of 15–100 MΩ.
Experimental protocol
Trigeminal responses were recorded within the TCC and in a separate set of experiments within the VPM, and challenged by topiramate intravenously, or locally by microiontophoresis directly on second-order or third-order neurons respectively. Recorded neurons demonstrated a stable response to dural vessels stimulation. The amplitude of the evoked firing was estimated by the means of post-stimulus histograms after a series of 25 stimuli.
For each intravenous treatment, dural vessel stimulation-evoked firing was recorded before (three baselines) and 5, 10, 15, 20, 30, 40, 50, 60, 70, 80, and 90 minutes after topiramate (30 mg/kg in 0.3 ml of solution) or its vehicle control were injected. For the same time points, the mean firing rate of spontaneous activity before and after drug administration was also calculated. Infusion of topiramate or its vehicle control was over 1 minute to allow detection of any transient changes in arterial blood pressure.
For the iontophoretic studies, neurons were tested for a stable response to electrical stimulation of dural vessels and an increased firing rate to microintophorized glutamate receptor agonists. Three baseline responses to dural vessels stimulation were collected 5 minutes apart. Each one of the glutamate receptor agonists tested for each experiment was ejected for 4–6 seconds, with retaining period of 40 seconds to avoid glutamate receptor desensitization. Once five stable baseline cycles of the glutamate receptor agonist-evoked responses were recorded, topiramate or vehicle control were ejected (75–95 nA) for 3–4 minutes over glutamate receptor agonist cycles. At the conclusion of topiramate or vehicle control ejection, agonists’ cycles were continued for 15–30 minutes and the neuronal responses were observed. To test the local effect of topiramate antagonism over normal baseline transmission, the mean firing rate of spontaneous activity over 150 seconds was calculated and compared to the mean spontaneous firing before, during, and after microiontophoresis of topiramate.
The location of the recording site was obtained by either direct marking of the site by ejection of charged pontamine sky blue at 4 µA for 10 minutes via a BAB-350 pump (Kation), or by reconstruction from a marked reference point and microdrive readings. Upon termination of the experiment, the brain and spinal cord were removed and processed for histological verification using the brain atlas by Paxinos and Watson (38).
Data analysis
All statistical analyses were conducted using SPSS (version 17; IL, USA). For the microiontophoretic experiments, the mean net firing rate was calculated for each of the successive cycles of glutamate agonists during each of the test conditions. Five baseline pulses were analysed to avoid variations of the responses of a cell between individual pulses and the reliability of the measurements was tested using Cronbach's alpha. The average of the three baseline cell firing obtained following stimulation of dural vessels was used for further comparisons. A repeated measures ANOVA for responses evoked by glutamate agonists and dural stimulation or of spontaneous activity was computed with two factors, Drugs and Repeats, to determine intra- and inter-drug effects and interactions. Bonferroni corrections were applied and when the assumption of sphericity with regards to the factor of Repeats was violated adjustments were made for the degrees of freedom and p values according to the Greenhouse−Geisser correction. The results from all cells were analysed together and compared by a series of paired t-tests to examine the effect of each intervention in turn. Significance was assessed at the p < 0.05 level. To compare the effect of systemic administration of topiramate and its vehicle control the effects of the drugs over time and the interaction of the drug used and time were evaluated by comparing cell firing changes for each individual cell using an analysis of variance with repeated measures followed by Bonferroni tests as described above. All data were converted as a percentage of the average of the baseline responses and are expressed as the mean value and the standard error of the mean (SEM) for each treatment group.
Results
A total of 35 wide dynamic range neurons (n = 21 animals) were studied in the TCC (Figure 1A). Calculations of neuronal conduction velocities were estimated by measuring the distance between the MMA stimulating site and the cervico-medullary recording site. An additional 1 ms was included for the central synaptic delay (40). All neurons tested demonstrated increased probability of firing of A-fiber inputs in response to electrical stimulation of the MMA, with an average latency of 8 ± 1 ms to onset (Figure 1B). Neurons firing with such latency were classified as receiving inputs from Aδ-fiber. Of those neurons 37 % additionally demonstrated C-fiber inputs with an average latency of 35 ± 2 ms. All cells were characterized as WDR and displayed convergent inputs from the ophthalmic cutaneous dermatome of the head (Figure 1E).
Summary of the location and characterization of trigeminovascular neurons. (A) Summary of the locations and a representative example of trigeminocervical complex (TCC) dorsal horn recording sites of nociceptive neurons receiving convergent synaptic input from the dura mater. Filled circles indicate the locations of the neurons that were marked by ejection of pontamine sky blue and open circles indicate the location of neurons reconstructed according to microdriver references. (B) An example of a second-order neuron firing in response to dural vessels electrical stimulation. (C) Summary of the locations and a representative example of recording sites within the ventroposteromedial thalamic nucleus (VPM). Filled circles indicate the locations of the neurons that were recovered by ejection of pontamine sky blue and open circles indicate the location of neurons reconstructed according to microdriver references. Locations are plotted on ideal cross-sections. (D) An example of third-order neuron firing in response to dural vessels electrical stimulation. (E) All cells recorded in the TCC displayed convergent inputs from the ophthalmic cutaneous dermatome of the head (V1) and neurons recorded in the VPM displayed convergent inputs from the first (V1) and second division (V2) of the trigeminal nerve. (F) Action potentials generated in response to N-methyl-
In the VPM, a total of 34 cells (n = 22 animals) responding to trigeminovascular activation were studied, were mostly located along the shell region of the VPM with the posterior complex and the ventroposterolateral thalamus (Figure 1C). Cells responded with an increased probability of firing to electrical stimulation of the SSS and the main body of response was found to occur with an average latency of 11 ± 0.5 ms to the onset, classified as Aδ-fiber activity (Figure 1D). C-fiber activity was recorded in 50% of the thalamicneurons with an average latency of 35 ms. Characterization of the receptive field was used to classify 11/34 neurons as NS and 23/34 as WDR. The units displayed convergent trigeminal viscerosomatic inputs with cutaneous receptive fields located on the contralateral craniofacial region of the ophthalmic and maxillary trigeminal divisions (Figure 1E).
To be included in the study, a probability of neuronal firing of greater than 30% was required and overall the recorded cells in the current study demonstrated a probability of firing of 53 ± 5%. Only cell bodies were recorded, which were characterized by their biphasic action and the increased firing in response to glutamate receptor agonists (Figure 1F).
Effects of topiramate in the trigeminocervical complex
Effects of intravenous topiramate on trigeminocervical neuronal firing
Topiramate administrated intravenously at 30 mg/kg significantly inhibited dural-evoked firing in response to Aδ-fiber activity (n = 8; F3,18 = 4.71, p < 0.05; Figure 2A) by a maximum of 36 ± 6% 60 minutes following administration (t13 = 3.82, p < 0.005). At the 90-minute time point no recovery of cell firing in response to MMA stimulation was seen. C-fiber responses and spontaneous neuronal activity from second-order neurons were not significantly affected (p ≥ 0.31; Figure 2B, C). Intravenous administration of dH2O given as vehicle control and had no effect on Aδ- or C-fiber firing or on spontaneous activity (n = 7; p ≥ 0.47; Figure 2). In all animals topiramate had no significant effects on blood pressure: mean ± SEM systolic blood pressure 5 minutes pre-topiramate administration, 115 ± 16 mmHg vs. 110 ± 18 mmHg 5 minutes post-topiramate administration, respectively.
Effect of intravenously administrated topiramate on responses of trigeminocervical neurons. (A) Topiramate at 30 mg/kg produced a long lasting inhibition of ∼30% of Aδ-fiber input on second-order neurons. (B−C) Although a small inhibitory action of topiramate was seen on C-fiber input (B) and spontaneous activity (C) of second-order neurons, this effect was not significant along the cohort.
Effects of microiontophoretic administration of topiramate on trigeminocervical neuronal firing
Local microiontophoretic application of topiramate (80–95 nA) on second-order neurons over a period of 3–4 minutes significantly attenuated dural-evoked firing corresponding to Aδ-fiber inputs by 32 ± 7% (n = 10; t9 = 3.54, p < 0.05; Figure 3A,B). Following a 15-minute recovery period, cell firing was only recovered to 89% compared to baseline levels. C-fiber activity in response to MMA stimulation was significantly inhibited by 35 ± 4% (n = 5; t4 = 4.13, p < 0.05; Figure 3C). Spontaneous cell firing was not affected by microiontophorezed topiramate (p = 0.47; Figure 3D).
Effect of microiontophoretic application of topiramate on trigeminovascular responses of trigeminocervical neurons. (A,B) Local microinjection of topiramate directly on second-order neurons significantly attenuate firing in response of Aδ- (B) and C-fiber input. Trigeminovascular firing recovered within 15⊟30 minutes (C). Topiramate had no effect on spontaneous activity when ejected locally on trigeminocervical neurons (D).
Microiontophoretic ejection of control ions (OH−) had no effect on trigeminovascular activation (p ≥ 0.10).
Effects of microiontophoretic administration of topiramate on glutamate receptor agonist-evoked firing
Microiontophoretic application of topiramate (85–95 nA) was tested on NMDA, fluorowillardiine (AMPA receptor agonist) and iodowillardiine (GluK1 kainate receptor agonist) -evoked firing from second-order neurons in the TCC. For all three agonists tested all baseline responses were reliable (Cronbach's α ≥ 0.98) and there was no difference across the mean firing of the five repeated epochs recorded during baseline conditions (fluorowillardiine: F4,32 = 1.64, p = 0.19; iodowillardiine: F4,32 = 0.62, p = 0.65; NMDA: F1,10 = 0.38, p = 0.61). Iontophoretic currents for NMDA were 35–70 nA and for fluorowillardiine and iodowillardiine currents were kept below 25 nA.
Overall local injection of topiramate on second-order neurons by means of microiontophoresis significantly inhibited kainate receptor firing in response to iodowillardiine by 49 ± 11% (n = 9; t8 = 3.63, p < 0.05; Figure 4A). The sensitivity to topiramate did vary between neurons as 4/9 cells were inhibited only by 18 ± 5% and 5/9 cells were inhibited by 73 ± 9%. In half of the cells, recovery was recorded within a minute of topiramate microiontophoresis cessation and in the remained recovery was more long-lasting for up to 15 minutes. The recovery of iodowillardiine-evoked firing was independent to the percentage of inhibition during topiramate's ejection. Topiramate had no significant effects on either fluorowillardiine- (n = 9; Figure 4B) or NMDA-evoked firing (n = 8; Figure 4C) compared to control ions ejection (p < 0.72), although, in two out of the nine cells tested for AMPA receptor's responses, topiramate inhibited fluorowillardiine-evoked firing by 43%.
Effects of microiontophoretically delivered topiramate on the firing rates of second-order neurons to pulsed ejections of the glutamate receptor agonists iodowillardiine, fluorowillardiine, and N-methyl-
Ejection of control ions had no effect on either glutamate receptor agonist-evoked firing.
Effects of topiramate in the ventroposteromedial thalamic nucleus
Summary of the actions of topiramate within the trigeminocervical complex (TCC) and the ventroposteromedial thalamic nucleus (VPM) on spontaneous and evoked firing

NMDA, N-methyl-
Effects of intravenous topiramate on trigeminothalamic activity
Topiramate was administrated intravenously at the same dose (30 mg/kg) as in the TCC experimental group. Topiramate significantly inhibited trigeminovascular activity from third-order neurons in the VPM in response to Aδ-fiber activity, evoked following SSS stimulation (n = 8; F4,25 = 2.88, p < 0.05; Figure 5A), by a maximum of 29 ± 9% (t12 = 5.34, p < 0.005) 40 minutes following administration, without demonstrating any significant changes on blood pressure. Similar to the effects of the drug in the TCC, at the 90 minute time point full recovery of cell firing was not seen. Topiramate had no effect on spontaneous firing (F11,66 = 1.35, p = 0.30; Figure 5C) and on C-fiber-evoked firing, although a trend towards inhibition was recorded (F2.04 = 2.88, p = 0.15; Figure 5B).
Effect of intravenously administrated topiramate on responses of third-order neurons within the ventroposteromedial thalamic nucleus. (A) Topiramate at 30 mg/kg produced a long lasting inhibition of ∼30% of Aδ-fiber input on third-order neurons but had no effect on C-fiber-evoked firing (B) or spontaneous activity (C). Vehicle control had no effect on either trigeminovascular or spontaneous activity.
Administration of vehicle control had no effects on firing within the VPM (n = 6; p ≥ 0.31).
Effects of microiontophoretic administration of topiramate on trigeminothalamic activity
Microiontophoretic application of topiramate (75–90 nA) significantly inhibited Aδ-fiber-evoked firing in response to SSS stimulation by 21 ± 5% (n = 10; t9 = 3.54, p < 0.05; Figure 6A) and firing was recovered to 82% (t8 = 3.134, p < 0.05) 15 minutes post cessation of ejection compared to baseline levels. Full recovery was recorded within 30 minutes.
Effect of microiontophoretic application of topiramate on trigeminovascular responses of third-order neurons. Local microinjection of topiramate directly on third-order neurons significantly attenuate firing in response of Aδ-fiber (A) but had no effect on C-fiber firing (B) or on spontaneous neuronal activity (C).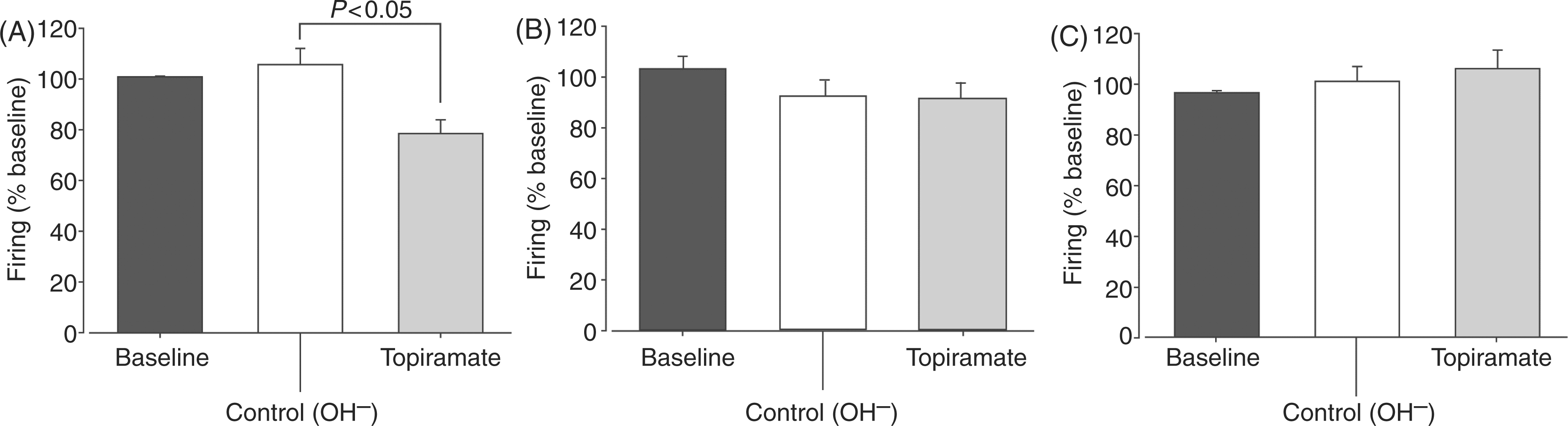
Microiontophoretic ejection of control ions (OH⊟) had no effect on trigeminovascular activation (t8 = 0.23, p = 0.83). Neither topiramate nor control ions had any effect on C-fibers activation (p ≥ 0.49; Figure 6B) or on thalamic spontaneous activity (p ≥ 0.48; Figure 6C).
Effects of microiontophoretic administration of topiramate on glutamate receptor agonist-evoked firing
For all three agonists tested there was no difference across the mean firing of the five repeated epochs recorded during baseline and all baseline responses were reliable (Cronbach's α ≥ 0.99). Iontophoretic currents were the same as in the TCC experimental group. Topiramate (80–95 nA) had no effect on NMDA-evoked firing (n = 9; t8 = 0.47, p = 0.65; Figure 7C) and induced a small but non-significant inhibition of fluorowillardiine-evoked firing compared to the effects of control ejection (n = 9; t8 = 9.33, p = 0.38; Figure 7B). As in the TCC, a small number of ventroposteromedial thalamic neurons (3/9) responding to fluorowillardiine demonstrated a small inhibition in the range of 22–24% in response to topiramate ejection. Iodowillardiine-evoked firing in the VPM was significantly inhibited by topiramate by 43 ± 7% (n = 9; t8 = 4.94, p < 0.005; Figure 7A) and this was not different from the effects of topiramate on kainate receptors activation in the TCC experimental group (p = 0.69). The sensitivity to topiramate did vary between the neurons; one-third of the cells were inhibited by 32 ± 11% and the rest were inhibited by 65 ± 3%. Similar to responses recorded within the TCC, recovery was either immediately or delayed over several minutes post cessation of ejection.
Effects of microiontophoretically delivered topiramate on the firing rates of second-order neurons to pulsed ejections of the glutamate receptor agonists iodowillardiine, fluorowillardiine, and N-methyl-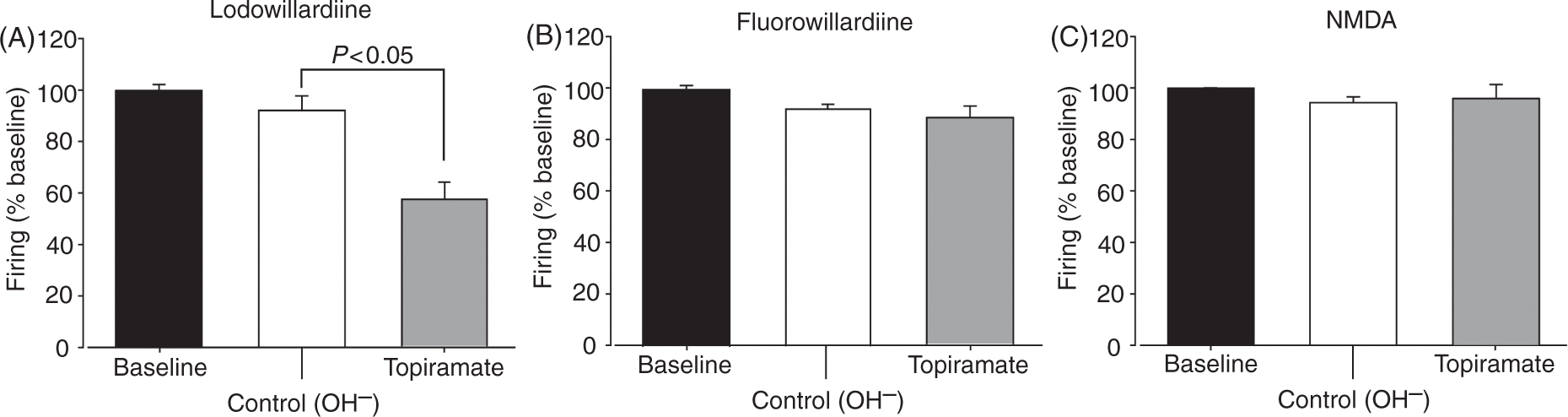
Control ions had no effect on either glutamate receptor agonist-evoked firing.
Discussion
The data presented point to a dominant effect of GluK1 kainate receptor antagonism as the significant glutamatergic mechanism for the migraine preventive effect of topiramate. Inhibition was seen after intravenous administration and crucially after microiontophoretic administration both at the level of the trigeminocervical complex and in the sensory thalamus. The data do not preclude involvement of other receptors nor do they address if longer periods of administration might have even more dramatic effects. The essential purpose of the study was to identify and support novel targets for preventive medicine development. The data do suggest that a pure GluK1 kainate receptor antagonist merits consideration for study as an anti-migraine preventive.
A pivotal pathway for migraine pathophysiology is the trigeminovascular input from the dural vessels, via the trigeminal ganglion, to the TCC, which is the key relay centre for the transmission of nociceptive information to higher brain centres where pain sensation is perceived (36). Second-order neurons from the TCC project to the thalamus and the VPM is the principal thalamic relay conveying nociceptive information from the meninges to the somatosensory cortex. The importance of the thalamus in migraine pathophysiology is clearly evident in brain imaging studies that show consistent thalamic activation in spontaneous attacks of migraine (41). The thalamus receives additional interest as auditory and visual information is also relayed through thalamic nuclei and it could be invoked to explain the sensitivity to light, sound, and non-cranial allodynia that accompany migraine attacks (42). There is increasing data demonstrating that drugs with established efficacy in the acute or preventive treatment of migraine can modulate trigeminovascular activity within thalamic nuclei (43–45). In the current study topiramate was effective in attenuating trigeminovascular activity via both systemic and microiontophoretic administration consistent with direct anti-nociceptive effects on at least two sites in the trigeminovascular pathway.
Glutamatergic mechanisms in migraine neurobiology have received increasing interest as glutamate is a key neurotransmitter responsible for the transmission of nociceptive information along the trigeminothalamic pathway. Genetic studies have demonstrated a strong link between an excess availability of glutamate in the brain and the genetic susceptibility to the development of migraine (32,46). Glutamate is strongly implicated in elements of the pathophysiology of the disorder, including trigeminovascular activation, central sensitization and subsequent development of allodynia, and cortical spreading depression. The presence of glutamate in the transmission of sensory information implicates the involvement of glutamate receptors in the modulation of trigeminovascular activation in key brain areas involved in the pathophysiology of migraine. Thus glutamate receptor antagonists could potentially be used as innovative neuronally targeted treatments of migraine (35).
Topiramate and kainate receptors in migraine pathophysiology
Topiramate has been suggested to display its antiepileptic effects via multiple mechanisms (25,27,31) including blockade of glutamate-related excitatory neurotransmission. Specifically, a number of studies have demonstrated that topiramate inhibits excitatory responses elicited by selective activation of the kainate receptor in the hippocampus, basolateral amygdala, cortex, and cerebellum (28,29,47,48). The kainate receptor is a member of the ionotropic glutamate receptors family with five known subunits GluK1–5, former GluR5−7 and KA1 and 2 (39). In vivo, topiramate selectively blocks kainate receptor activation-induced seizures, but not seizures induced by AMPA or NMDA (49,50). Interestingly, the effect of topiramate on kainate receptors appears to be GluK1 dependent, as the drug had no effect on in vitro preparations of GluK2 (former GluR6) kainate receptors (51). A partial action of topiramate has been also observed on AMPA receptors; however, in agreement with the current study, topiramate's action on this receptor had lower efficacy, while the blockade of kainate-evoked currents occurs at concentrations within the free serum levels of this anticonvulsant in patients (28,29,52).
The pharmacological mechanisms accounting for the anti-migraine efficacy of topiramate have not hitherto been systematically investigated, although the drug was found effective in a number of preclinical studies using animal models of migraine (53–55). In our study, topiramate selectively attenuated the excitability of trigeminovascularly linked neurons in response to GluK1 receptors activation by ∼50% in both the TCC and the VPM. It is thus very likely the inhibition of trigeminovascular activity seen by local application of topiramate both at the level of the TCC and the thalamus is driven, at least partially, by blockade of GluK1 carrying kainate receptors. As inhibition of trigeminovascular firing by microiontophoresis of topiramate in both nuclei was long lasting, it is more likely that, at least in some cells, this inhibitory action could be driven by the long-lasting inhibition of kainate receptor-evoked firing seen in 50% of the neurons. Blockade of kainate receptors could additionally contribute to the suppressive effects of topiramate on trigeminovascular activity both in the TCC and the VPM when applied systemically. It is exciting from a pharmacogenetic perspective that a recent study identified polymorphisms in the GRIK4 gene, one of the genes encoding kainate receptor subunits, that are associated with outcome in the preventive treatment of migraine with topiramate (56). The lack of selective pharmacological tools has hampered the study of the GluK4 subunit in detail. Using in situ hybridization Bahn and colleagues (57) demonstrated that GluK4 and 5 subunits coexist with functioning GluK1–3 subunits in the same population of cells, suggesting that the native kainate receptors may be heteromeric tetramers composed of different combinations of dimmers of these subunits. In our study, although iodowillardiine has a selective affinity for the GluK1 subunit, but not for GluK2 or 3 subunits, it is likely that the drug could also act on GluK1/GluK4 assemblies present within the TCC and VPM.
Kainate receptors are a promising target given the pathophysiology of migraine, as they are not as widely distributed within the CNS as other glutamate receptors. They are certainly found in the trigeminal ganglia (58,59) and in structures of the ascending migraine pathway, such as the TCC (60), the thalamus (61), and the sensory cortex (62). In a human molecular genetic study of migraine, the 11q24 locus which maps, among other candidates, near the GRIK4 gene, which codes for the GluK4 kainate receptor subunit (former KA1), has demonstrated significant linkage to migraine with aura (63). Both pre-and post-synaptic GluK1 kainate receptors in the TCC are suggested to be involved in trigeminovascular nociceptive processing (64), and blockade of post-synaptic GluK1 receptors inhibits trigeminovascular activation (65,66). Local application of kainate GluK1 selective antagonists at the level of the VPM additionally attenuates trigeminothalamic activity, while interactions between kainate and GABAA and 5-HT1B receptors have further been recorded in this thalamic nucleus (61). Further to the pre-clinical studies demonstrating anti-nociceptive actions of kainate antagonists in animal models of trigeminovascular activation, small clinical trials have provided similar data.
The AMPA/GluK1 antagonist LY293558, tezampanel was effective in treating headache pain in two-thirds of migraineurs studied, and also relieved accompanying symptoms, such as nausea and photophobia (67). This compound was well tolerated by patients and has no vasoconstrictor liability (68). Similarly, in a randomized double-blind study, the GluK1 kainate antagonist LY466195 (69) was effective in relieving acute migraine (70) and further supports this thesis. Our current data further suggest a role in prevention for the kainate receptor. By acting centrally within the ascending trigeminothalamic pathway, topiramate partially inhibited excitatory neurotransmission via kainate receptors and thus attenuated trigeminovascular activity. A drug with similar but selective action of partial inhibition may be a distinct advantage in terms of avoiding unwanted side effects of broader glutamatergic inhibition (35).
Other potential mechanisms of action of topiramate in migraine
The current study investigated the pharmacological actions of topiramate restricted to ionotropic glutamate receptors along the ascending migraine pathway; other mechanisms may be involved. Intravenous application of topiramate at the same dose used in the current study, was previously found to block cortical spreading depression, which is believed to be the underlying mechanism of migraine aura (54). We have previously shown that blockade of kainate receptors with LY466195, at doses that have no actions on NMDA receptors, is not sufficient to block cortical spreading depression (65). In the current study in half the neurons recorded both in the TCC and the VPM, the topiramate-evoked inhibition of kainate receptor activation induced-firing recovered soon after cessation of topiramate's ejection and this cannot easily explain the inhibition of trigeminovascular activity over several minutes. It is thus likely other pharmacological mechanisms of action contribute to the inhibitory effects of topiramate on trigeminovascular activity. Interestingly, an in vitro study using neurons from the cortex and periaqueductal grey, areas that play a major role in the pathophysiology of migraine, demonstrated that topiramate blocked high-voltage-activated Ca2+ channels (71). A number of studies have additionally shown that topiramate can act on trigeminal neurons and their fibers at the periphery and block the release of calcitonin gene-related peptide (55), which has been associated with migraine attacks. This mechanism of action might again be associated with blockade of Ca2+ channels as they mediate the release of both glutamate and calcitonin gene-related peptide from trigeminal neurons (72).
Methodological considerations
The use of microiontophoresis in the current study has considerable advantages in terms of the application of drugs in neuroanatomically well-defined brain areas, allowing the demonstration that topiramate is capable of acting on second- and third-order neurons in the TCC and VPM, respectively. Microiontophoresis was additionally a useful neuropharmacological tool in terms of agonist−antagonist pairing and facilitated the actions of topiramate on kainate but not AMPA or NMDA receptors. A limitation of this technique has been the significant failure rate due to blockage of topiramate-filled barrels. Thus, for each setup more than one barrel was used for topiramate and consequently not all ideal comparisons were made in all neurons. It is also not possible to know the concentration of drug that each cell is exposed to (73), and this can be a limitation when trying to establish if the observed effects would occur at clinically relevant doses. Nevertheless, microiontophoresis remains a valuable tool to dissect the neuropharmacology of distinct brain regions and groups of neurons.
The effect of topiramate may differ when administered systematically, as it is not possible to localize its effects. This is particularly a problem for all thalamic effects that are also seen at the level of the TCC, as the effect could be additive or, indeed, entirely due to the TCC effect. One might thus expect a more prominent action of topiramate in the VPM when administrated intravenously compared to the ∼30% inhibition recorded at the TCC level. This might suggest topiramate has a much higher potency and efficacy in blocking trigeminovascular nociceptive processing at the level of the TCC compared to third-order neurons in the VPM. In support of this conclusion, local application of topiramate attenuated C-fibers activity within the TCC but not within the VPM nucleus. An additional aspect to consider is that using microiontophoretic application of topiramate at such a local scale, the drug may be acting post-synaptically, pre-synaptically, or at both sites, depending on the synaptic location of the receptor under target. As GluK1 receptors are found both on primary trigeminal afferents (both A- and C-fibers) and on second-order neurons within the TCC, topiramate may target both sites, and thus have more prominent actions on both types of fibers, compared to VPM where GluK1 receptors are located mostly post-synaptically. Failure to record full recovery of cell firing in response to dural stimulation 90 minutes following intravenous administration of topiramate might reflect a prolonged activity of topiramate as the compound's half life has been shown to be approximately 21 hours (74) or of a local effect due to limited local diffusion.
Another important issue is that only acute trigeminal nociception was studied. As topiramate is used as a preventive, its longer term effects need consideration. Nevertheless, electrical stimulation of the dura/vessel complex has been a highly predictive model (75), even for migraine preventives (45), probably due to the restricted nature of the phenotype of dural stimulation in humans (76).
Conclusions
Topiramate can modulate trigeminovascular nociceptive transmission in the trigeminothalamic pathway. Inhibition of trigeminovascular neurons activated by a nociceptive stimulus has been predictive of anti-migraine activity. A major pharmacological site of action of topiramate in both the TCC and the VPM nucleus is the kainate receptor. This pharmacological action may explain some part of its efficacy in migraine prevention and further suggests a possible role of kainate receptors in migraine pathophysiology. Understanding the mechanism of action and the pharmacology of topiramate may help to develop new medications that are specifically designed for migraine prevention.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.