
Abstract
Introduction: Chronic daily headache (CDH) represents a temporal profile of headache (15+ days/month; 4+ hours/day; >6 months). We report the first comprehensive and largest levetiracetam (LEV) trial in CDH.
Methods: A 27-week, multi-centre, randomised, placebo-controlled, cross-over, phase III B study assessed efficacy of a target of 3 g/day LEV of 6 placebo tablets/day in CDH. Primary efficacy was headache-free rate (HFR) while secondary parameters were loss of diagnostic criteria; severity; duration; disability; associated features; pain; and quality of life.
Results: Ninety-six patients were recruited (baseline HFR 10.4 ± 14.6%; median 0%). At onset of history 73 (74.1%) had migraine +/− aura and 35 (36.5%) had tension-type headache (TTH). Over the six months preceding recruitment 54 (56.3%) had migraine and 42 (43.8%) had TTH. Headache history was 22.6 ± 15.0 years (median 20.0). Eighty-eight received placebo and 89 received LEV with >80 receiving stable dose in either arm. LEV achieved 3.9% increased HFR over placebo, showing a trend but not significance. There was 9.9% increase in loss of CDH diagnostic criteria re: headache days/month for LEV over placebo (p = .0325), reduced disability (p = .0487) and reduced pain severity for LEV (p = .0162). The Short-Form Quality of Life assessment instrument (SF-36) showed impaired mental health on LEV (p = .001).
Discussion: These findings conflict with reports of LEV efficacy, mandating placebo control in headache trials. Primary efficacy equated to one extra headache-free day/month with reduced disability and pain intensity. Mental health was reduced on LEV. The 10% loss of diagnostic criteria, decreased intensity and disability suggest a subpopulation with CDH where LEV remains a therapeutic option.
Introduction
Chronic daily headache (CDH) describes a temporal profile in which headaches occur on at least 15 days per month, lasting at least four hours for at least six months (1–3). CDH is not a specific headache type and may include chronic migraine, chronic tension-type headache, hemicrania continua and new daily persistent headache. CDH affects approximately 4–5% of the population (4) but accounts for more than 40% of presentations to specialist headache clinics (5).
Levetiracetam (LEV) is an anti-epileptic medication (AEM) with a distinct chemical structure and pharmacokinetic profile (6,7). It is a pyrrolidone derivative (8), acting as a synaptic vesical protein-binder (9). LEV demonstrated pain relief efficacy in animal experiments (10,11) and has psychotropic properties (12).
CDH is often resistant to prophylaxis although some efficacy has been demonstrated in clinical trials with AEMs, including valproate (13,14), gabapentin (15) and topiramate (16,17). There are some small uncontrolled trials of LEV suggesting benefit in migraine. It follows that LEV may prove efficacious in the prophylaxis of CDH.
This paper reports the findings of the first comprehensive and largest randomised, double-blind, placebo-controlled trial of LEV in CDH.
Methods
Design
This was a 27-week, multi-centre, randomised, double-blind, placebo-controlled, cross-over phase IIIB study of LEV in CDH prophylaxis (Figure 1). Randomisation was external to participating centres, according to standardised operating procedures, generating randomisation codes using SAS. The study was approved by each centre’s human research ethics committee, acknowledging this was an off-license use of LEV, approved in Australia for refractory epilepsy.
Protocol design for the randomised, placebo-controlled, cross-over study of levetiracetam.
Based on a previous study of gabapentin in CDH (15), a difference of 7.5% in the primary efficacy variable (headache-free rate [HFR]) represents a clinically relevant response in this resistant headache form and was adopted for the current study. A mean difference of 7.5% would be detected at the 5% level with 80% power if at least 70 subjects were enrolled. Allowing for attrition, a target of 120 patients was selected. Patients were recruited from both hospital neurology clinics and neurologists’ private practices.
At the screening visit (Week-4) patients were assessed for inclusion and exclusion criteria and written informed consent was obtained. Medical history (including prior medications) was obtained, and general physical and neurological examinations were performed. Headache types, at the onset of the headache history and in the six months prior to recruitment, were defined by the principal neurologist at each site. Laboratory tests, including haematology, serum creatinine, liver function and pregnancy tests were performed.
Visual analogue scale (VAS) assessment of the overall headache intensity during the preceding week (0 = no headache to 10 = worst possible headache) and Short Form-36 (SF-36) Quality of Life survey were completed. Patients were given headache diaries to record: daily headache severity (coded from 0 = no headaches experienced through to 5 = very severe headaches); headache duration; degree of disability (coded from A = no disability through to E = total disability); use of concomitant medication; and associated features including nausea, vomiting, photophobia, phonophobia and aggravation by movement.
Inclusion criteria comprised adults 18–65 years; capacity to consent; headache history confirming CDH; females of child-rearing potential practicing reliable contraception with negative screening pregnancy test; ability to comply with the protocol; and capacity to follow instructions.
Exclusion criteria included hypersensitivity to LEV; previous treatment with LEV; confounding neurological or general medical disorders; treatment with other experimental drugs within one month of baseline; history of illicit drugs or alcohol abuse during the previous year; liver function tests more than twice the upper limit of normal; severe renal impairment; consumption of more than 4 mg of ergotamine per week, more than 300 mg of sumatriptan per week or 15 mg of zolmitriptan or naratriptan per week; administration of parenteral opioids more than once per month or the use of oral opioids (except for codeine-containing analgesics) on average more than once per month over the last six months; and patients with post-traumatic headaches.
Patients using other headache prophylactic medications were required to maintain stable doses for the duration of the trial.
Target dose for up-titration was 6 tablets of LEV (3000 mg/day) or 6 tablets of matching placebo. Patients commenced either 500 mg LEV or placebo, one at night, for three days. The dosage increased every fourth day by one tablet to 3 tablets twice daily. A reduction from 2500 mg to 2000 mg or 3000 mg to 2500 mg was accepted when intolerable side effects occurred. Once the maximum tolerated dose was achieved, stable therapy was maintained for six weeks. Thereafter, a two-week down-titration commenced, followed by a one-week washout and cross-over to the other treatment arm, adopting an identical regimen (Figure 1).
At Visit 1 (Week 0), baseline headache diaries were evaluated, VAS completed and adverse events (AEs) and concurrent medications recorded. Patients satisfying ongoing inclusion criteria were randomised to either LEV or placebo.
At Visit 2 (Week 9) and Visit 3 (Week 12), daily headache diaries were collected, VAS completed, concurrent medications and AEs recorded. At Visit 3, medication for the second treatment period was dispensed and pregnancy tests repeated.
At Visits 0, 2 and 4, the SF-36 Quality of Life survey was administered. At Visits 2 and 4, physical and neurological examinations were performed and the physician’s and patient’s global assessment of response recorded.
At the conclusion of the second treatment arm, there was a two-week down-titration, a four-week washout and the final visit, Visit 5, 27 weeks after randomisation. Physical and neurological examinations were repeated, pregnancy tests conducted, concurrent medications reviewed, AEs noted, daily headache diaries collected and VAS completed. Patients were asked to identify which treatment arm was active.
Efficacy and safety variables
Primary efficacy
The primary efficacy measure was HFR for each stable treatment period calculated from the daily headache diary (DHD).

Secondary efficacy
Secondary efficacy parameters included loss of CDH diagnostic criteria, headache severity, headache duration, disability, associated features, VAS and Quality of Life (SF-36).
Safety
Safety variables included nature, frequency, severity and relationship to study medication of all AEs; neurological and physical examinations; and laboratory results.
Stastistical analysis
Primary efficacy analysis
Primary efficacy analysis was performed on HFR (and repeated for data that included only headaches of at least four hours’ duration).
The outcome for each individual in the treatment period was the HFR during stable dosing. This was expressed as percentage of headache-free days (R) out of total days on stable dose of medication (N). This fixed-effects model includes terms for treatment, treatment period and patient identification.
Where data were not normally distributed, a ranked analysis of covariance (ANCOVA) was performed in which the dependent variable was the difference in HFR between treatment and placebo.
Secondary efficacy analysis
Diagnostic criteria for CDH were compared between treatment arms using a McNemar’s paired test for categorical data. Headache severity, duration and overall severity scores (severity × duration) were compared using a fixed-effects model. When residuals were non-normal, a ranked ANCOVA was adopted. Mean severity was the outcome of interest and the model included terms for treatment and treatment period. Pre-treatment mean headache severity was included as a baseline covariate.
Degree of disability was recorded on an ordinal scale from A (absence of disability) to E (totally disabled). Results were coded as A = 0 to E = 4 and summarised using descriptive statistics. Data were analysed using a fixed-effects model in the same way as headache severity, subject to the assumptions of the model.
Associated headache features were nausea, vomiting, photophobia and phonophobia, headache aggravated by movement and whether other headache medications were taken. These were presented as a percentage of diarised headache days, summarised by treatment and tested for each type of disability, using a fixed-effects model with the outcome of interest being the percentage of days with the associated feature as a proportion of the number of days during stable dosing.
VAS scores evaluated perception of headache intensity over the week prior to assessment, represented by a vertical stroke on a line depicting headache severity from 0 (no headache) to 10 (most severe headache). The distance along the line was summarised and tabulated by treatment arm with change from baseline. The difference between treatments in VAS was tested using a fixed-effects model with the VAS as the outcome and treatment, visit, patient and baseline (screening visit) as covariates.
Results
Between 28 May 2004 and 18 April 2007, seven sites recruited 96 subjects representing the safety population. They were aged 48.8 ± 12.5 years (range 18–65 years, median 46.4) of whom 95% were Caucasian and 53% female. They consumed 3.3 ± 2.01 cups of caffeine-containing drinks per day (range 0–12, median 3) and 4.2 ± 6.9 standard alcoholic drinks per week (range 0–42, median 1). Hours of sleep per night were 6.6 ± 1.62 (range 2–10, median 7). The HFR, expressed as a percentage of days free of headache, was a baseline of 10.4 ± 14.6% (range 0–50%, median 0%), indicating a recalcitrant headache profile, with the majority having headaches every day. Duration of headache experience prior to inclusion in the study was 22.6 ± 15.04 years (range 1–57, median 20.0).
Reason for withdrawal from treatment at withdrawal (safety population)
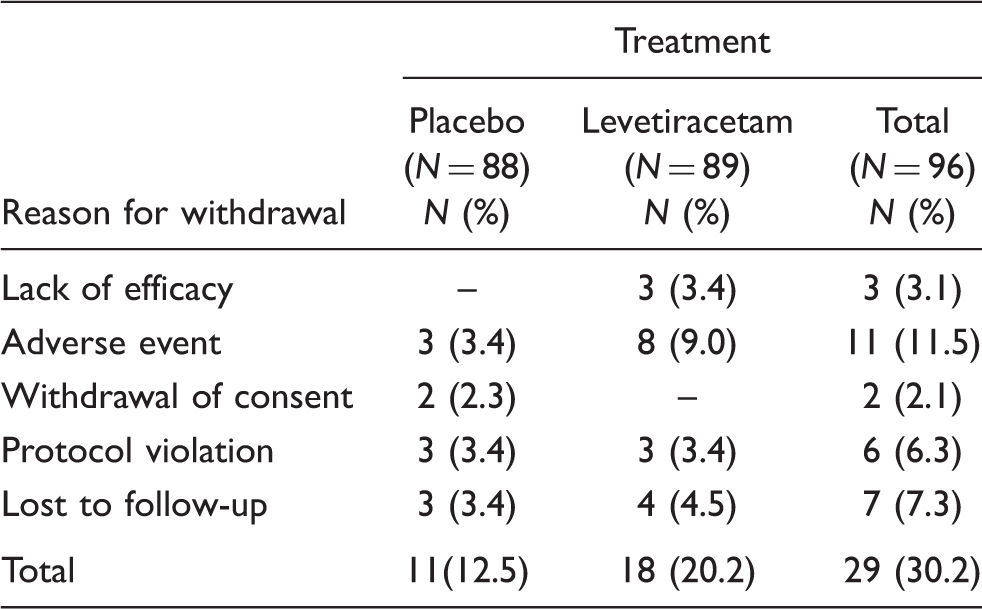
Neurologists considered that 50 (52.1%) had migraine without aura; 23 (24.0%) had migraine with aura and 35 (36.5%) had tension-type headaches at the onset of headache history. Ten (10.4%) had new daily persistent headache, and 2 (2.1%) had primary hemicrania continua at the onset. No patients had cluster, primary stabbing, primary cough, primary exertional, primary sexual, primary hypnic or primary thunderclap headaches at the onset of the headache history.
During the six months prior to inclusion, neurologists diagnosed 35 (36.5%) with migraine without aura; 19 (19.8%) with migraine with aura; and 42 (43.8%) with tension-type headaches. Forty (41.7%) had daily persistent headaches unclassifiable using the International Headache Society (IHS) classification (18). A single patient (1.0%) had primary hemicrania continua and no patients had cluster, primary stabbing, primary cough, primary exertional, primary sexual, primary hypnic or primary thunderclap headaches.
Primary efficacy
Headache-free rate (%) by treatment (efficacy population)

SD = standard deviation.
The HFR, for headaches lasting at least four hours, was 24.67% on LEV and 22.67% on placebo. This favoured LEV but was not statistically significant (p = .1904).
Secondary efficacy
Loss of diagnostic criteria for chronic daily headache, by treatment (efficacy population)

CDH = chronic daily headache. HPM = headache days/month.
Other secondary efficacy criteria
There was no significant difference between the two arms with respect to headache duration, attack severity, degree of disability or presence of associated symptoms (nausea, vomiting, phonophobia, photophobia or aggravation of headache by movement). There was also no difference in use of concomitant medications in headache management.
A separate analysis of degree of disability was performed using only headache-days during the stable-dose period (headache severity > 0). The mean degree of disability on placebo was 1.34, whereas on LEV it was 1.25. This favours LEV, indicating that the headaches were less disruptive of daily life (p = .0487). The mean percentage of headaches featuring light/sound sensitivity was higher (31.06%) on LEV than on placebo (28.19%). The difference was significant (p = .0409).
The VAS data show a lower mean score for subjects receiving LEV (4.99) compared with placebo (6.11), indicating lower headache intensity on LEV (p = .0162).
Patient’s global impression of change
Patients’ global impression of improvement or deterioration and treatment arm was analysed using the Cochran-Mantel-Haenszel test, and showed no correlation (p = .5254).
Patient identification of treatment arm
Of the 79 patients who completed post-study Visit 5, only 33 (41.7%) correctly identified the treatment arms.
Quality of life
Only the mental health domain of the SF-36 showed significant change, favouring placebo over LEV (61.05 vs, 56.5; p = .0011).
Physical examination
There was no difference demonstrated between LEV and placebo in clinical examination.
Concomitant medication
There was no difference between LEV and placebo for use of concomitant medications.
Adverse events
In the pre-randomisation period, 45 AEs were reported. During stable dosing, 100 AEs occurred on placebo (44 considered possibly, probably or definitely related to study medication). On LEV, 177 AEs were reported (81 considered possibly, probably or definitely due to study medication).
Four patients on LEV reported serious AEs (one severe hemiplegic migraine and three surgical intervention unrelated to study medication). No serious AEs occurred on placebo.
There were six severe AEs on placebo and 14 on LEV, none considered to be consequent to study medication. Of the remaining AEs, the only apparent differences were dizziness (1 placebo vs, 7 LEV) and lethargy (0 placebo vs, 3 on LEV).
Overall, no action was required for AEs in 54/100 on placebo and 107/177 on LEV.
Discussion
Several AEMs have shown efficacy in headache prophylaxis (13–17,19). Small prospective open-label and retrospective trials suggest a role for LEV in migraine therapy (20–26). These studies lacked placebo control and had small patient populations. Only one targeted difficult-to-treat migraines (25), evaluating LEV in transformed migraine. Our paper reports the first comprehensive and the largest randomised, placebo-controlled, double-blind trial of LEV in the treatment of headache focusing upon the most refractory form, CDH. It is also the largest trial of LEV in headache.
Headache type at the onset of headache history and over the six months preceding entry into the study was identified according to IHS classification (18). Most (74.1%) had migraine (with or without aura) and/or tension-type headache (36.5%) at onset of headache history. There were fewer migraineurs during the six months preceding study enrolment (56.3% migraine, with or without aura, and 43.8% tension-type headaches). This may reflect a natural history of longstanding headache with metamorphosis of migraine to a form indistinguishable from tension-type headache. Alternatively, it may relate to difficulty identifying headache type once frequency becomes almost daily.
The methodology adopted for this study mirrored that used in the Australian multi-centre placebo-controlled gabapentin trial in CDH (15), employing the same sample size and primary and secondary efficacy criteria. Acute medication overuse was an exclusion criterion, removing it as a confounding variable. The definition of medication overuse employed in this study identified absolute quantities of medication, rather than the number of doses, as the former better reflects the quantity of medication exposure. This definition is more precise than that which has been advocated by the IHS (18).
For primary efficacy, LEV achieved a 3.9% increase in HFR over placebo, demonstrating a trend but failing statistical significance. Among secondary efficacy variables, loss of diagnostic criteria for CDH regarding headache days per month, showed a 9.9% increase for LEV over placebo (p = .0325). There was reduced disability on LEV over placebo (p = .0487). Headache intensity, using VAS, was also reduced on LEV (p = .0162).
The 3.9% HFR difference equates to one extra headache-free day per month over placebo, a questionable benefit. However, headache intensity is reduced by approximately 10% and with reduced headache-related disability.
Despite reduced headache severity, based on both VAS and disability scales, there was an increase of photophobia/phonophobia on LEV (p = .0409). This appears contradictory, as photophobia/phonophobia occur during more severe headaches. Either LEV potentiated these symptoms or it was ineffective in alleviating them, rendering them apparent with decreased headache intensity.
Behavioural disorders have been reported on LEV (27,28). Impaired mental health was also found in this study, using SF-36 (p = .001).
These data raise doubts regarding reported efficacy of LEV in small prospective open-label and retrospective studies (19–26). Most of these assessed LEV in episodic migraine rather than CDH. One study reported substantial benefit with LEV in transformed migraine, with quoted mean headache frequency of 24.9 days per month, satisfying one of the two CDH criteria. In the current study, approximately 60% satisfied IHS criteria for migraine (18), suggesting that the conflicting results are unlikely to be explained by differences in population selection. The fundamental difference between the current study and those previously reported is the presence of a placebo control.
The placebo effect is quite complex (29) and in headache trials, in particular, its contribution can be a major confounder (30,31). Patients may report AEs on placebo (32), and three patients in the current study withdrew on that basis. Furthermore, the HFR improved by 50% from baseline on placebo (10.4% to 15.5%) and almost 100% on LEV (10.4% to 19.1%) but still failed to achieve statistical significance because placebo, rather than baseline, was the comparator. In uncontrolled trials reporting efficacy, it was the change from baseline which was considered. It follows that the previously reported efficacy may represent a placebo effect. This emphasises the need for a placebo arm in all headache trials.
To put this trial into context, it has to be appreciated that the study population had a median duration of headache history of 20 years (range 1–57), during which numerous headache remedies had failed. Further, the median HFR was 0% (range 0–50), indicating that the majority of subjects experienced headaches every day. These data must influence expectations of therapy, rendering more modest gains acceptable. Although this was a negative study in terms of the primary efficacy criterion, the fact that almost 10% more of the study population lost CDH diagnostic criteria on the basis of headache days per month whilst on LEV suggests that there may have been a sub-population in which significant reductions in headache days was realised. This, together with the demonstrated reduction in headache intensity and disability, indicates that LEV should still remain an option in this highly resistant headache population when other treatments have failed.
Footnotes
Financial disclosure
This was an investigator-initiated study funded by an educational grant supplied by UCB Pharma, which also supplied study medication and matching placebo. UCB Pharma had no control of the data or input into the analysis or writing of this paper. RG Beran has served on an advisory board for levetiracetam in the treatment of epilepsy. PJ Spira has no financial disclosure to make.