
Abstract
Introduction: Intranasal sumatriptan is an option for the treatment of migraine; however, nasal delivery using conventional spray pumps is suboptimal.
Methods: Adult subjects (n = 117) with migraine were enrolled in a multicentre, randomised, double-blind, parallel group, placebo-controlled study. A single migraine attack was treated in-clinic with sumatriptan 10 mg, sumatriptan 20 mg or placebo administered intranasally by a novel bi-directional powder delivery device when migraine was moderate or severe.
Results: A greater proportion of subjects who received sumatriptan were pain-free at 120 minutes compared with those who received placebo (10 mg/20 mg sumatriptan vs. placebo = 54%/57% vs. 25%, P < .05). Significant benefits were also observed for pain relief at 120 minutes (84%/80% vs. 44%, P < .001/.01) and as early as 60 minutes (73%/74% vs. 38%, P < .01) and for 48 hours sustained pain-free (P < .05). Treatment-related adverse events were rare, with a metallic taste being the most commonly reported (10%/13%).
Conclusions: Sumatriptan nasal powder administered using the new device during a migraine attack was effective and well tolerated.
Introduction
Migraine is a recurrent headache of moderate-to-severe intensity that is associated with gastrointestinal, neurological and autonomic symptoms. During migraine attacks, gastric emptying is delayed, and as a consequence oral treatments may have a delayed onset of effect. Furthermore, patients may suffer from nausea and vomiting, which may make taking an oral tablet difficult. Intranasal formulations of some triptans, including sumatriptan, have been developed with the intent of achieving a faster onset of action, while avoiding the need to inject the drug. Clinical studies have shown that intranasal sumatriptan delivered by conventional liquid spray pumps is an effective means of relieving migraine (1–3).
A recent review of the pharmacokinetics and clinical effects of different sumatriptan formulations describes the tradeoff between fast onset offered by subcutaneous (SC) injection compared with the more favorable adverse-event profile and patient acceptability offered by oral and nasal sumatriptan (4). For patients, a barrier exists related to difficulties using the conventional nasal devices, which may also cause variability in the clinical effects (5). However, intranasal delivery is considered the most attractive route for patients with nausea and/or vomiting, particularly if the speed of onset and reliability can be improved (4).
Current nasal sprays deposit the drug mainly in the non-ciliated part of the nasal passage anterior to the nasal valve. To avoid drip-out, the patient typically sniffs, which rapidly moves the concentrated drug along the floor of the nose to the oral cavity, often resulting in a bitter taste (3). The bi-directional delivery system used in the powder delivery device (OptiNose) has documented significantly improved deposition to the respiratory and olfactory mucosa (6), which is essential for rapid systemic absorption and for potential direct transport from the nose to the brain (N2B) (5). Bi-directional delivery uses two aspects of nasal anatomy to improve the extent and reproducibility of dosing while avoiding the risk of lung inhalation (6,7). First, during exhalation against a resistance the soft palate closes due to positive pressure, separating the nasal and oral cavities. Consequently, it becomes possible to use smaller particles in a nasal spray and avoid lung deposition by exhalation through the mouth during nasal administration. Second, during closure of the soft palate there is a communication pathway between the two nostrils, located behind the wall separating the two passages. Under these circumstances, it is possible for air flow to enter via one nostril and leave by the other. Indeed, a recent phase I study with the same device and sumatriptan formulation used in this study showed a faster and more extensive systemic absorption across the nasal mucosa than the existing sumatriptan liquid spray pump, and also suggests some degree of N2B transport or local action via the olfactory and trigeminal nerves, which innervate the nasal mucosa (8).
The aim of this study was to evaluate the efficacy and safety of two doses of a powder formulation of sumatriptan delivered intranasally with the novel device in comparison with placebo.
Methods
Study design
This randomised, double-blind, placebo-controlled, dose-ranging, parallel group study enrolled adult subjects with a history of migraine headache at 10 centres in the Czech Republic. Nine centres were in hospital neurology clinics, and one centre was a private clinic. All subjects gave written informed consent to participate in the study, which was conducted in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice. The study was approved by both a central ethics committee and local ethics committees at each site.
Inclusion criteria were age 18–65 years; a developing or established attack of migraine with or without aura according to the International Headache Society (IHS) criteria, of moderate (grade 2) or severe (grade 3) intensity and no improvement in the attack at the time of assessment; migraine present for at least one year, with a three-month, well-documented retrospective history; 48 hours of freedom from headache between attacks of migraine; age at first diagnosis of migraine less than 50 years; an average of ≥1 and ≤6 migraine attacks per month for the past six months; reporting the migraine attack and attending the clinic within four hours of the onset of the attack; verified air flow through both nostrils and an ability to close the soft palate; and the ability to trigger the breath-actuation mechanism of a device in accordance with the instructions for use. Exclusion criteria included an inability to distinguish other headaches from migraine; other headaches at a frequency of more than six days per month; subjects who use drugs excessively for headache (i.e. who use medication for acute headache more than 10 days each month); resistance to migraine drugs; use of ergotamine, ergot-type medications or any 5-HT1 agonist or narcotic analgesics within the previous 24 hours before treatment; current use of drugs for migraine prophylaxis; use of any analgesic within 12 hours before treatment; treatment with monoamine oxidase A (MAO-A) inhibitors or attending for treatment within two weeks of discontinuation of MAO-A inhibitor therapy; treatment with selective serotonin-reuptake inhibitors; using a decongestant within six hours of attendance at the clinic on the treatment day; hemiplegic or basilar migraine; a history, symptoms or signs of ischemic cardiac, cerebrovascular or peripheral vascular syndromes or uncontrolled hypertension (systolic blood pressure >140 mmHg and/or diastolic blood pressure >95 mmHg); known nasal obstruction due to nasal deviations, polyposis, severe mucosal swelling or any other reason; current uncontrolled nasopharyngeal illness; known velum insufficiency; and extensive nasal and/or sinus surgery.
At the screening visit, eligible subjects underwent a physical examination, 12-lead electrocardiogram (ECG), measurement of vital signs after the subject had been sitting for five minutes and blood and urine sampling for clinical laboratory tests. Nasal patency and ability to close the soft palate were checked. Each subject was trained in the use of the device, and their ability to achieve a suitable outflow was established. Subjects were instructed to telephone the clinic at the onset of their first migraine attack.
There was a single treatment day. Subjects were transported to the clinic as soon
as they reported a migraine attack. Continued compliance with
inclusion/exclusion criteria was checked. A pregnancy test was conducted on all
female subjects of childbearing potential. The subjects' vital signs were
recorded after they had been sitting for five minutes, a 12-lead ECG was
recorded and blood and urine samples taken for safety evaluation. Baseline
details of each subject’s migraine attack were recorded. The
subjects were instructed again in the use of the device and then randomly
allocated to treatment, either 10 mg sumatriptan, 20 mg
sumatriptan or placebo. Efficacy and safety assessments were made for 120
minutes after administration. The OptiNose powder device is shown in Figure 1. Prior to each
administration, the capsule inside the device was punctured by pressing the
piercing button once. The subject placed the sealing nosepiece into the nostril
selected for administration and the mouthpiece into their mouth. To effect the
administration, the subject took a deep breath with the mouth open, then closed
the lips around the mouthpiece and exhaled into the device, delivering the
powdered drug into the nose via the nosepiece. For the 10 mg dose,
one device was provided containing the 10 mg dose and the subject
performed the administration to one nostril. For the 20 mg dose, two
devices were provided, each containing a 10 mg dose and the subject
performed the administration to both nostrils, in rapid succession. In the
placebo group, one-half of the subjects administered one placebo device and
one-half of the subjects administered two placebo devices (one to each nostril)
to maintain blinding in the same manner. The devices were identical in
appearance and therefore neither the study staff nor the study subjects were
aware of whether active treatment or placebo was being administered. Where the
treatment consisted of a single device (active or placebo), the administration
was to the nostril on the same (ipsilateral) side as the migraine headache
occurred. If the headache was bilateral, the single administration could be made
to either side. The administration(s) were supervised by the investigator or a
designee. The OptiNose powder delivery device.
Each capsule contained the active ingredient sumatriptan succinate at a dose of 10 mg base equivalent (14 mg of the succinate salt) with a mean particle size of 15 µ. The device was provided pre-filled. The residuals of sumatriptan in the capsules were measured using a high-performance liquid chromatography method. The placebo device was identical in external appearance to the active device, but contained an empty capsule.
Efficacy assessments
On attending the clinic, patients had the pre-treatment details of the migraine attack recorded. Headache severity score (0 = no pain, 1 = mild pain, 2 = moderate pain, 3 = severe pain); the level of functional disability (0 = no disability, able to function normally; 1 = performance of daily activities mildly impaired, can still do everything but with difficulty; 2 = performance of daily activities moderately impaired, unable to do some things; 3 = performance of daily activities severely impaired, cannot do all or most things, bed rest may be necessary); and migraine-associated symptoms (nausea, vomiting, phonophobia, photophobia) were recorded immediately prior to dosing and 15, 30, 60, 90 and 120 minutes post-dose. Subjects measured time to meaningful relief using a stopwatch. Meaningful relief was defined subjectively by the patient. If the subject was not pain-free at the 120-minute assessment, the subject was permitted to take rescue medication. The sustained pain-free (SPF) status (defined as pain-free within 120 minutes with no use of rescue medication or relapse within 48 hours) of the subject was recorded at the follow-up visit.
Safety assessments
Safety assessments included adverse events, laboratory tests, vital signs, 12-lead ECG and physical examination. Details of all reported adverse events were recorded throughout the study, with severity graded as mild, moderate or severe, and a relationship to treatment assigned based on the judgment of the investigator. Blood and urine sampling for laboratory tests and recording of a 12-lead ECG were performed at screening, before dosing on the treatment visit and at follow-up. The following haematological parameters were assessed: haemoglobin concentration, total red blood cell count, erythrocyte sedimentation rate, mean corpuscular volume, total white cell count, neutrophils, lymphocytes, eosinophils, monocytes, basophils and platelets. The following blood biochemical parameters were assessed: hepatic function (total bilirubin, alkaline phosphatase, gamma-glutamyltransferase, aspartate transferase, alanine transferase), renal function (urea, creatinine) and clinical chemistry (sodium, potassium, calcium, albumin, total protein, glucose). Urine analysis consisted of pH, glucose, protein, occult blood, ketones, specific gravity, bilirubin and urobilinogen. Measurement of vital signs (blood pressure and pulse rate) and physical examination were performed at screening and follow-up. In addition, vital signs were measured prior to treatment and after 15, 30, 60, 90 and 120 minutes on the treatment day.
Statistical analysis
It was initially planned that a modified intent-to-treat analysis would be performed on all subjects who presented with a migraine, received study medication and had a post-dose primary efficacy evaluation. A review of blinded data listings prior to database lock found that 13 subjects were major protocol violators: 12 subjects with a headache severity score of 1 (mild) at baseline on the treatment day and one subject who received rescue medication within 120 minutes of study treatment administration. Therefore a per-protocol analysis excluding these major violators was included in the statistical analysis plan. This change to the planned analysis was made prior to database lock and unblinding of the study. This paper presents the results for the per-protocol population.
All statistical tests were performed using SAS version 8.2 software (SAS Institute Inc., USA). The level of significance, alpha (α), for this study was 0.05. The primary end point was the proportion of subjects pain-free at 120 minutes after treatment. Secondary end points were relief of headache (defined as a decrease in headache intensity from severe [grade 3] or moderate [grade 2] to mild [grade 1] or none [grade 0]), time to meaningful relief, level of functional disability, incidence of associated symptoms (nausea, vomiting, photophobia phonophobia), need for rescue medication and SPF status. Calculation of the recently introduced end point, sustained pain-free and no adverse events (SPFNAE), encompassing a combination of parameters of what the patients desire of their medication for migraines, was also performed, using SPFNAE = SPF(1-AE) (9).
The primary efficacy analysis was Fisher’s exact test comparing the proportion of subjects treated with sumatriptan in each dose group who were pain-free at 120 minutes post-dose to those on placebo who were pain-free at 120 minutes. Fisher’s exact test was also used to capture differences between each sumatriptan group and the placebo group at different time points for relief of headache, incidence of associated symptoms, rescue medication use and SPF status. Time to meaningful relief was estimated using Kaplan-Meier survival analysis and analysed using log-rank test for treatment difference between the sumatriptan groups and the placebo group. Data were censored at 120 minutes post-dose. The level of functional disability, measured at 15, 30, 60, 90 and 120 minutes, was analysed using log-linear models.
Sample size calculations were based on proportions of subjects pain-free at 120 minutes of 11% for placebo and 41% for sumatriptan 20 mg, as predicted from published studies (1,2). A sample size of 40 subjects per treatment group allowed 80% power to detect this difference at the 5% level (two-sided).
Results
Subject characteristics
Subjects were recruited from April 2007 to September 2007. A total of 117
subjects (100 females, 17 males) were randomised to treatment. Demographics and
baseline characteristics of treated migraines were similar among all treatment
groups (Table 1).
Apart from one subject in the placebo group who was lost to follow-up, all
subjects completed the study. All subjects were included in the safety
population. One subject in the 10 mg sumatriptan group, four
subjects in the 20 mg sumatriptan group and seven subjects in the
placebo group had only mild headache on treatment. In addition, one subject in
the 10 mg group took rescue medication within 120 minutes of dosing.
All these subjects were major protocol violators and were excluded from the
per-protocol population. Figure
2 presents a flow diagram showing the progress of subjects through
the study, including the numbers analysed. All treatment administrations were
made in the clinic in the presence of the investigator or designee, and
compliance was 100%. An audible rattle (due to the capsule in the
device spinning around) was recorded on each administration made, indicating
that each device had actuated correctly. There were no device failures, and the
majority of patients found the powder device easy and intuitive to use despite
only one rehearsal before self-administration during the moderate-to-severe
migraine attack. Measurements of residuals showed that a mean ±
standard deviation (SD) of
77 ± 11% was emitted from the
device, yielding a mean delivered sumatriptan dose of 7.7 mg and
15.4 mg, respectively, from the 10 mg and
20 mg nominal doses. Disposition of subjects and numbers analysed. Demographics, migraine history and characteristics of pre-treatment
headache A subject may have had more than one of the listed symptoms.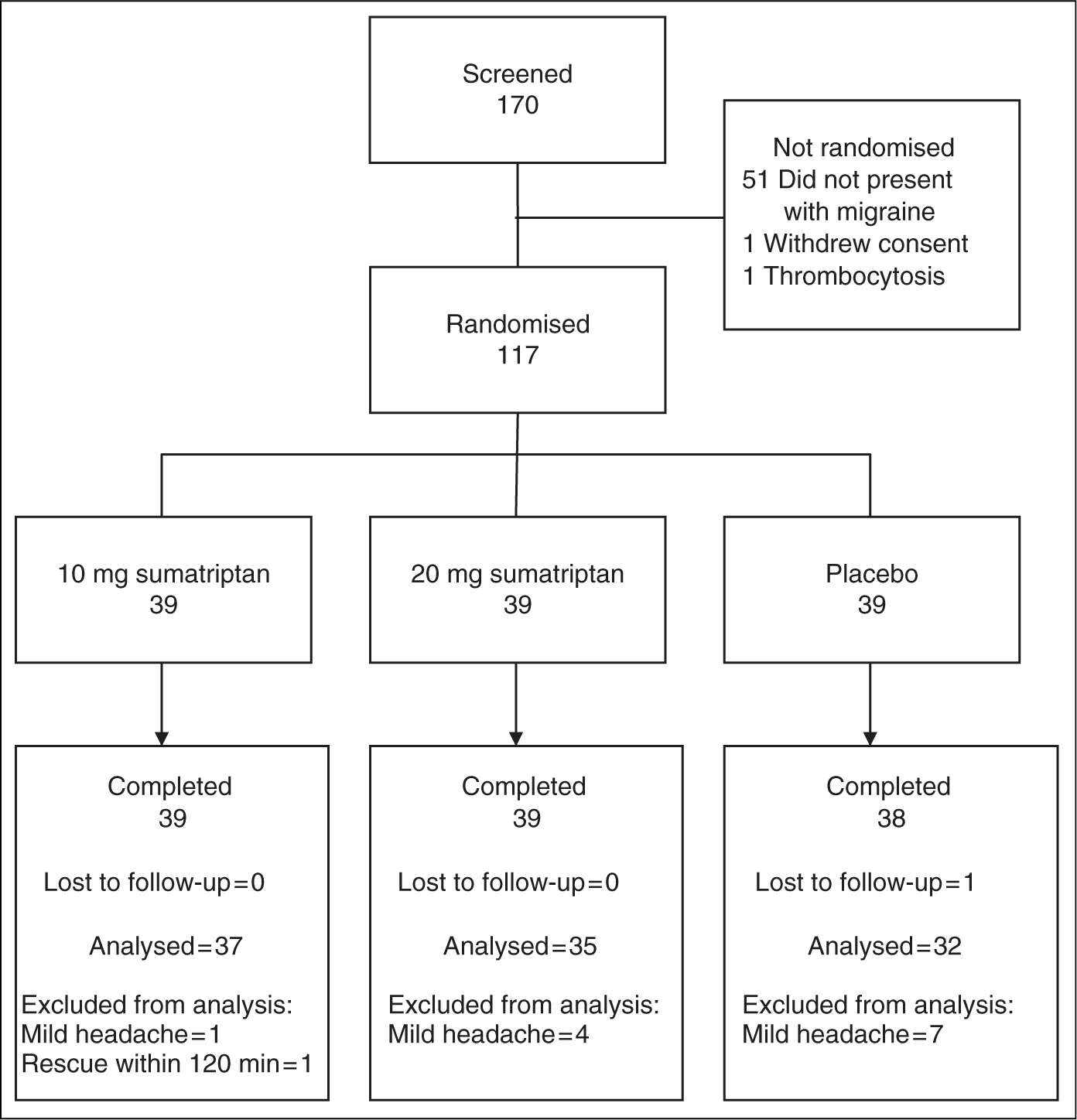

Efficacy
A significantly greater proportion of subjects in both the 10 mg
sumatriptan (54% vs. 25%;
P < .05) and
20 mg sumatriptan (57% vs. 25%;
P < .05) groups were
pain-free at 120 minutes compared with placebo (Figure 3). For both the 10 mg
and 20 mg sumatriptan doses, the proportion of subjects with relief
of headache was significantly greater than placebo at 60 minutes
(10 mg 73% vs. 38%,
P < .01; 20 mg
74% vs. 38%,
P < .01), 90 minutes
(10 mg 78% vs. 41%,
P < .01; 20 mg
77% vs. 41%,
P < .01) and 120 minutes
(10 mg 84% vs. 44%,
P < .001; 20 mg
80% vs. 44%,
P < .01) post-dose (Figure 4). Proportion of subjects pain-free at 120 minutes after treatment.
P < .05
vs. placebo using Fisher’s exact test. Proportion of subjects with relief of headache at various time points
after treatment. Relief of headache is defined as a decrease in
headache intensity from severe (grade 3) or moderate (grade 2) to
mild (grade 1) or none (grade 0). **P < .01,
***P < .001
vs. placebo using Fisher’s exact test. The median time to meaningful relief was 54 minutes for 10 mg
sumatriptan (P < .05) and
50 minutes for 20 mg sumatriptan
(P < .05), with both times
significantly faster than the median time of 120 minutes for placebo (data for
the survival analysis were censored at 120 minutes and therefore a median time
longer than 120 minutes for the placebo group was not possible). A significantly
greater proportion of subjects in both the 10 mg sumatriptan
(76% vs. 44%;
P < .05) and
20 mg sumatriptan (71% vs. 44%;
P < .05) groups had
meaningful relief compared with placebo. At baseline, the majority of subjects had daily activities moderately or severely
impaired (see Table
1). At 90 minutes post-dose, no disability was reported in
60% of subjects in the 10 mg group and 54%
in the 20 mg group compared with 31% in the placebo
group (P < .05). Similar
differences were observed at 120 minutes post-dose (10 mg
65% vs. 31%,
P < .05; 20 mg
63% vs. 31%,
P < .05). Additionally, at
60 minutes post-dose, 46% of subjects in the 10 mg
sumatriptan group had no disability compared with 25% in the placebo
group (P < .05). There were marked reductions in the incidence of nausea, photophobia and
phonophobia compared to baseline in both the 10 mg and
20 mg sumatriptan groups between 60 and 120 minutes post-dose. For
the 10 mg sumatriptan group, the incidence of phonophobia was
significantly lower than placebo at 90 minutes (13% vs.
34%; P < .05)
and the incidence of photophobia was significantly lower than placebo at both 90
minutes and 120 minutes post-dose (16% vs. 41%;
P < .05). The
reductions in the incidence of phonophobia or photophobia for 20 mg
sumatriptan were not, however, significantly lower than placebo at any time
point. For nausea, the reductions were not statistically significant compared
with placebo for either the 10 mg or the 20 mg dose. The
incidence of vomiting was very low in all three treatment groups, and no
significant treatment effects were observed. The proportion of subjects who required rescue medication at 120 minutes
post-dose was significantly lower in the 10 mg sumatriptan group
compared with placebo (11% vs. 38%;
P < .05). A smaller,
non-significant reduction was observed between the 20 mg sumatriptan
group and placebo (26% vs. 38%). A significantly greater proportion of subjects were SPF from 120 minutes up to 48
hours post-dose (European Medicines Agency “Guideline on the
clinical investigation of medicinal products for treatment of
migraine”) (10) in both the 10 mg (47% vs. 22%;
P < .05) and
20 mg (49% vs. 22%;
P < .05) sumatriptan groups
compared with placebo (Figure
5). Proportion of subjects sustained pain-free up to 48 hours after
treatment. Sustained pain-free is defined as pain-free
within 120 minutes with no use of rescue medication or relapse
within 48 hours.
P < .05
vs. placebo using Fisher’s exact test. The SPFNAE calculated from the SFP and AE rates were 39% and
37% for 10 mg and 20 mg sumatriptan,
respectively. In total, seven subjects (18%) in the 10 mg sumatriptan
group, nine subjects (23%) in the 20 mg sumatriptan
group and two subjects (5%) in the placebo group experienced at
least one adverse event. The incidence of treatment-related adverse events was
higher in both sumatriptan groups than in the placebo group (six subjects
[15%] in both the 10 mg and 20 mg
sumatriptan groups vs. one subject [3%] in the placebo group). The
most common adverse event following administration of 10 mg or
20 mg sumatriptan was dysgeusia, reported as a bitter or metallic
taste, in 10% subjects receiving the 10 mg dose and
13% receiving the 20 mg dose. There were no cases of
chest discomfort or pain, paresthesia or asthenia in the active treatment
groups. One subject in the placebo group experienced oral paresthesia. No
adverse events resulted in the withdrawal of a subject from the study. There
were no serious adverse events in the study. There were no clinically
significant findings in relation to vital signs, 12-lead ECG or clinical
laboratory test results for any of the treatment groups. Sumatriptan powder in 10 mg and 20 mg doses administered
intranasally using the bi-directional delivery device was highly effective in
treating a single migraine attack. The proportions of subjects pain-free were 54% for 10 mg
and 57% for 20 mg sumatriptan compared to
26–42% at two hours post-dose for the liquid sumatriptan
nasal spray (11) and
zolmitriptan nasal spray (35.6%) (12). The mean pain-free rate for 12
studies with the 100 mg oral dose was 28%, which is very
similar to the pain-free rate for six oral triptans of 29% (11,13) and the new
calcitonin gene-related peptide (CGRP) antagonist (14,15). The proportions of subjects with
headache relief at two hours were 84% and 80% for the
10 mg and 20 mg treatments, respectively. Headache
relief rates ranging from 50–78% have been reported for
the liquid 20 mg sumatriptan nasal spray, oral 100 mg
sumatriptan, the sumatriptan-naproxen combination, the new CGRP-antagonist and
other oral triptans (11,13,15,16).
As described in the statistical analysis, the current study used a per-protocol
analysis, excluding major protocol violators (mostly subjects with a mild [grade
1] headache at baseline); caution should therefore be exercised when
interpreting the comparative data with earlier studies, which may have handled
protocol violators differently. The placebo effect in the present study for both pain relief (44%)
and pain freedom (25%) is high, reducing the two-hour therapeutic
gain (TG) for pain relief to 40% and 36% and to
29% and 32% for pain freedom for 10 and
20 mg OptiNose powder, respectively. Similar placebo rates of
42–46% at two hours have, however, been reported for
pain relief in studies with nasal sprays
(placebo = 42%,
TG = 32%) (11), the new rapid-dissolving
formulation of sumatriptan
(placebo = 46%,
TG = 26%) (17) and the CGRP antagonist telcegepant
(placebo = 46.3%,
TG = 21.7%) (14). Despite lower
pain-free placebo rates at two hours for the conventional triptan nasal sprays
(4,12), the CGRP
antagonist (15) and
the sumatriptan-naproxen combination (16), ranging from
8–18%, these treatments also have lower pain-free TG
rates than the OptiNose powder device. The TG rates for the highest doses of
oral triptans were lower or similar to the OptiNose powder device (11,13). Only SC injection
of sumatriptan has a higher TG than the OptiNose powder device for pain relief
at two hours (placebo = 19%,
TG = 50%) (11). Higher patient expectancy for new formulations or technologies may be one factor
explaining the high placebo rates in studies with new formulations such as the
rapidly dissolving sumatriptan tablet (17), the new CGRP antagonist (14) and our new
sumatriptan powder drug-device combination. Many studies fail to show significant improvement at time points earlier than two
hours, and data on one hour are often not reported. In our relatively small
phase II study, the one-hour pain relief rates for 10 mg and
20 mg doses of 73%
(TG = 36%) and 74%
(TG = 37%) are higher than the
published data on sumatriptan oral 100 mg tablets, the oral
sumatriptan rapidly dissolving tablets and the 20 mg sumatriptan
nasal spray with relief rates of 49–52%
(TG = 20–22%) (11,16,17). This is achieved
with a sumatriptan dose more than 10 times lower than the oral
100 mg tablet and less than one-half of the marketed
20 mg sumatriptan nasal spray. The 6 mg SC injection is
the only formulation with similar one-hour pain relief rates of
69–73% (11). SC injection shows better TG at
one and two hours, but the Cmax is almost 10 times higher that of
10 mg OptiNose sumatriptan powder (8,11). Despite the 48-hour time interval in the current study, the SPF rates for both
10 mg (47%,
TG = 26%) and 20 mg
(49%, TG = 27%)
sumatriptan groups are high. For the 100 mg oral, 20 mg
nasal and 6 mg SC sumatriptan, the sumatriptan-naproxen combination
and nasal zolmitriptan 5 mg, the absolute SPF rates vary between
23% and 30% and the TG is at
15–20% (10,11,14). The absolute SPF rates of the
highest dose marketed oral triptans, vary between 19% and
26% (9).
It is possible that the differences between the present study and earlier
studies may be a reflection on how the data were collected. The earlier studies
collected data using diary cards, whereas the current study collected SPF data
at a visit to the clinic. The most common adverse event in the present study was bitter or metallic taste,
reported in four subjects (10%) and five subjects (13%)
in the 10 mg and 20 mg treatments groups, respectively.
Higher rates of bitter or bad taste have been reported following administration
of sumatriptan liquid spray by the nasal route, occurring in about
25% of patients receiving the 20 mg dose with a liquid
spray (3) and in
17.9% of patients using zolmitriptan nasal spray (12). When patients are
asked specifically about taste, the incidence of bitter taste with the
20 mg liquid nasal spray may be as high as 68% (12,18). A traditional
spray delivers most of the dose of the drug to the anterior, non-ciliated
segment and subjects commonly sniff to avoid drip-out, causing the concentrated
drug to be sucked posterior along the nasal floor to reach taste buds at the
base of the tongue, recording bitter taste. A broader mucosal distribution and
different clearance pattern will dilute the drug and minimise the amount
reaching the “bitter” taste buds at the base of the
tongue (6). Calculation of SPFNAE from a single, relatively small study must of course be
interpreted with great caution. Nevertheless, it is noteworthy that the SPFNAE
rates in the present study of 39% and 37% for
10 mg and 20 mg nasal, respectively, of OptiNose
sumatriptan are three times higher than the median value reported in a large
meta-analysis which included the six major marketed oral triptans (median
12%, range 7–22%) (9). It has been suggested that it is the rate of absorption, not the extent, which
explains the onset of action of triptans (19). The superior efficacy of
subcutaneous versus oral administration of sumatriptan and naratriptan was also
considered most likely due to a quicker rise in blood concentrations following
subcutaneous administration (20). Delivery of sumatriptan powder by the OptiNose device provides
more rapid and extensive direct absorption across the nasal mucosa to the blood,
resulting in a much earlier median tmax at 20 minutes compared to a
median tmax of 1.5 hours for the marketed nasal sumatriptan spray
(8,21). The fraction
absorbed from the nose following delivery with the doses and device used in this
study is estimated to be 30% compared to 10% for the
marketed sumatriptan spray, resulting in a more pronounced, two-tiered
absorption pattern with initial rapid nasal absorption and a slower delayed
gastrointestinal absorption (3,4,8,21). The comparable efficacy of the
10 mg and 20 mg doses in our study with the same
tmax but different cmax (8) also supports the hypothesis that
rate of absorption is more important than extent of absorption, but these
observations may simply reflect a dose response ceiling for delivery of nasal
sumatriptan powder by the bi-directional delivery device. The broader distribution of the sumatriptan powder formulation following
bi-directional delivery to the nasal mucosa may not only increase systemic
absorption compared with conventional liquid sprays, but potentially could also
result in enhanced direct or reflex-mediated actions of nasally delivered
sumatriptan on the trigeminal nerve, the ganglion and associated cerebral
structures involved in the pathogenesis of migraine (5,6,8). This provides an alternative
explanation for high efficacy despite low systemic exposure and the similar
efficacy of the 10 mg dose delivered to the side of the migraine as
20 mg divided between the two nostrils. However, the present study
design does not allow for any conclusions, and a more comprehensive discussion
of the potential role of N2B transport mechanism or direct actions on or via the
trigeminal nerve and ganglion is outside the scope of this paper. Convenience mostly drives the preference of patients for tablets, whereas the
speed of onset and the efficacy are the key drivers for preference for the nasal
spray. Among patients having tried two oral (regular tablet and fast-dissolving
tablet) and the nasal zolmitriptan formulation, as many as 40%
preferred the nasal zolmitriptan spray (22). Thus, it is likely that the faster
onset of action, higher rate of sustained pain freedom and lower rate of adverse
events with low rate of bitter taste will make users more attracted to the new
sumatriptan powder device combination. Sumatriptan at doses of 10 mg or 20 mg administered using the
new bi-directional powder delivery device was effective, safe and well tolerated in
treating a single migraine attack. The performance of the new device was good and
there were no device failures. The Czech Migraine Investigators Group are Petr Dočekal (principal
investigator, General Faculty Hospital, Prague), Luisa Bartlova (Motol University
Hospital, Prague), Edvard Ehler (Regional Hospital, Pardubice), Petr
Kaňovsky (University Hospital, Olomouc), Otaker Keller (Thomayer
University Hospital, Prague), Pavel Kunc (Private Clinic, Dobruška), Jan
Rejda (Hospital Most, Most), Ivan Rektor (St Ann’s University Hospital,
Brno), Stanislav Vohanka (Brno University Hospital, Brno) and Milena Zdvorila
(Hospital Chomutov, Chomutov). We thank Iveta Prosova, MD, for assistance in the
study. We also thank Graeme Hewson, Tony Flint and Colin Sheldrake for their
assistance in preparing the manuscript.


Safety
Discussion
Pain freedom and pain relief
The placebo effect
Early onset
Sustained pain freedom
Adverse events
Sustained pain-free plus no adverse events
Mechanisms of action
Conclusions
Footnotes
Acknowledgements
References