
Abstract
Introduction: Migraine pain is thought to involve an increase in trigeminal nerve terminal activity around large cerebral and meningeal arteries, leading to vasodilatation. Because prostaglandin E2 (PGE2) is elevated in cephalic venous blood during migraine attacks, and is also capable of inducing headache in healthy volunteers, we hypothesize that PGE2 dilatory receptors, EP2 and EP4, mediate the response.
Materials and methods: By the use of specific agonists and antagonists, the dilatory effect of PGE2 was characterized in rat cranial arteries by use of in vivo and in vitro methods. Furthermore, EP2 and EP4 quantitative messenger RNA (mRNA) receptor expression was studied in the rat craniovascular system.
Results: Our results suggest that EP4, and to a lesser degree EP2, receptors mediate the dilatory effect of PGE2 in the craniovascular system in rats. Thus, antagonism of these receptors might be of therapeutic relevance in migraine.
Keywords
Introduction
Prostaglandin E2 (PGE2) is a naturally occurring metabolite of arachidonic acid. PGE2 action is mediated through the distinct G-protein-coupled receptor family, EP, which is further divided into the subtypes EP1, EP2, EP3 and EP4 (1–3). The EP1 and EP3 receptors are functionally coupled to mobilization of Ca2+, whereas EP2 and EP4 receptors are coupled to Gs, causing stimulation of cyclic adenosine-3,5′-monophosphate (cAMP) formation (3). In vessels the functional effects of the EP1 and EP3 receptors are vasoconstriction, and EP2 and EP4 receptor activation causes vasodilatation (3,4).
One of many roles ascribed to PGE2 is in the pathophysiology of migraine. This neurovascular disorder is characterized by unilateral, pulsating and throbbing pain to the head (5). Numerous mechanisms are thought to be involved in headache pain, including dilatation of large cerebral and meningeal arteries, facilitating meningeal nociceptor activation, thereby resulting in pain (6). Various vasodilators, such as calcitonin gene–related peptide (CGRP) (7), glyceryl trinitrate (8) and pituitary adenylate cyclase–activating polypeptide (9), have been shown to reliably induce headaches in humans. In addition, CGRP receptor antagonists are effective against acute migraine (10,11).
A few studies have addressed the role of PGE2 in migraine, but its pharmacological mechanisms are yet to be delineated. PGE2 levels in jugular venous blood have been reported to increase significantly within one hour after onset of a migraine attack, peaking two to four hours after attack onset (12). Furthermore, increased levels of PGE2 have been detected in saliva (13,14) and blood (15) of migraineurs during attacks. In a recent study on healthy subjects, intravenous infusion of PGE2 induced headaches and a concomitant dilatation of the cerebral vessels (16). Furthermore, PGE2 is released from the dura mater encephali as a result of both electrical and chemical stimulation in vitro, indicating its role in nociceptor activation in rats (17,18). Indirect evidence of prostaglandin involvement in headache comes from the fact that nonsteroidal anti-inflammatory drugs, such as aspirin (19) and indomethacin (20), are commonly used in the treatment of acute headache. The effect of such drugs is to inhibit the cyclooxygenase enzymes and thereby prostaglandin synthesis. In addition, cAMP-coupled EP receptors in cultured rat trigeminal neurons release CGRP upon stimulation (21), thus indicating a role for PGE2 in the regulation of CGRP, a peptide implicated extensively in migraine pathogenesis. Nevertheless, it has not yet been deciphered which particular receptors are involved functionally or which of the receptor subtypes are expressed in the craniovascular system; this vascular circuit is pivotal in migraine etiology.
The aim of the present study was to investigate the hypothesis that PGE2 can dilate the rat craniovascular system using in vivo as well as in vitro preclinical models and that this response is mediated by EP2 and EP4 receptors.
Materials and methods
All experiments were performed according to the guidelines and regulations of the Danish Animal Experimentation Inspectorate (file: 2004/561–850) on the care and use of experimental animals. In all experiments male adult Sprague-Dawley rats were used (Taconic Europe, Ejby, Denmark). The rats were maintained in cages with a 12-hour light/dark cycle and had free access to food and water.
Improved ‘closed cranial window’ model
Forty rats (330–470 g) were anesthetized with pentobarbital (Mebumal® 65 mg kg−1) intraperoneally (i.p.), and depth of anesthesia was tested by suppression of the hind-paw reflex. Anesthesia was continuously supplemented with pentobarbital (Mebumal® 20 mg kg−1 h−1) intravenous (i.v.) during the experiment. The body temperature was maintained at 37.0 ± 0.5°C throughout the experiments using an automatic regulated heating plate (Letica HB101, Panlab, Barcelona, Spain). Following intubation the animal was mechanically ventilated by a respirator (SAR-830/AP, CWE, Ardmore, PA, USA) with 30/70% air mixture of O2/N2O, a stroke volume of 3.5–4.0 ml and a stroke rate of 55–65 per minute. The improved closed cranial window allows introduction of an indwelling catheter (Portex, Fine Bore Polythene Tubing, inner diameter 0.4 mm, Astratech, Taastrup, Denmark) glued to a tip (outer diameter 0.3 mm) in the right carotid artery for infusion of test substance instead of the femoral artery. As previously described (22), this improvement introduces the test molecule directly to the local cranial circulation, thus circumventing severe blood pressure drops. Other catheters were placed in the left and right femoral artery and vein for infusion of anesthetic, measurement of mean arterial blood pressure (MABP) and sampling of blood for gas tension analysis. Arterial blood samples were collected prior to, during, and at the end of each experiment, for analysis of partial pressure of oxygen (P aO2), carbon dioxide (P aCO2) and pH (ABL520, Radiometer, Brønshøj, Denmark). All values were kept within normal limits (pH 7.35–7.45, MABP 81.7–127.5 mmHg and P aCO2 35.2–42.7 mmHg). The closed cranial window model experiments were performed as described previously (22,23). In brief, the animal was placed in a stereotactic frame. Skin covering the dorsal surface of the skull was retracted and the connective tissue and muscle removed, leaving the right parietal bone exposed. The bone was thinned, making a window (10 × 7 mm2) by carefully drilling with a dental drill cooled by application of ice-cold synthetic interstitial fluid (SIF composition in mM: 108 NaCl, 3.48 KCl, 3.5 MgSO4, 26 NaHCO3, 11.7 NaH2PO4, 1.5 CaCl2, 9.6 sodium gluconate, 5.55 glucose and 7.6 sucrose; pH 7.4) until the middle meningeal artery (MMA) and/or the branch of the middle cerebral artery (MCA) were clearly visible while still leaving the skull intact. For visualization of the arteries, an intravital microscope (model MZ 16, Leica, Heerbrugg, Switzerland) was positioned above the cranial window. A video dimension analyzer (V94, Living Systems Instrumentation, USA) continuously monitored and measured the target artery diameter. MABP was measured constantly as well. All data were continuously displayed on a computer monitor using the data acquisition and analysis software Perisoft (version 2.0; Perimed AB, Järfälla, Sweden). At the end of the experiments, animals were sacrificed by an overdose of pentobarbital (150 mg kg−1 i.v.) or 1 M potassium chloride i.v.
Experimental design
The thinning of the skull induces mechanical stress; therefore, after surgery the animal was left to recover for at least 60 minutes. By then the artery was constricted and the baseline diameter was determined. In all experiments 150 µl of vehicle was infused intracarotid (i.c.) at the rate of 50 µl min−1. Each dose was constituted in a volume corresponding to one-eighth of the weight of the animal in order to minimize the injected volume (22). The dilatory capacity of the target artery was then tested with one single dose of CGRP (100 ng kg−1) (22). To study the dilatory effect on the MMA or MCA, doses of PGE2 and the EP2 agonists, butaprost (24) and ONO-AE1-329 (25,26), (at half-log increments) were administered via i.c. infusion or topically. The artery was allowed to return to baseline between each measurement. A submaximal dose of PGE2 (300 ng kg−1) was selected and repeated three times in order to confirm reproducibility of the response (Figure 1). In the case of antagonists, the percentage of inhibition was calculated by comparison between the first (before antagonist infusion) and the second dose of PGE2 (after antagonist infusion). The antagonists, AH6809 (EP2 antagonist) (1,27), BGC20-1531 (EP4 antagonist) (28) and SQ22536 (adenylate cyclase inhibitor) (29), were administered via i.c. infusion as follows: infusion of the antagonist for one minute followed by 100 µl saline over two minutes. Infusion of PGE2 for one minute was followed by 100 µl saline over two minutes. Changes in the MMA diameter and in MABP were measured one minute prior to the first infusion (baseline) and again after infusion of PGE2 (peak response of MMA).
A representative recording of the reproducible effect of three times PGE2 300 ng kg−1 on blood pressure (upper panel) and middle meningeal artery (MMA) (lower panel) after i.c. infusion for one minute (50 μl min−1) followed by saline.
In vitro studies
A total of 13 rats (350–450 g) were exsanguinated during CO2 anesthesia. The brains were removed and placed in chilled, oxygenated Ca2+-free Na+-Krebs buffer solution (in mM: 119 NaCl, 15 NaHCO3, 4.6 KCl, 0.026 EDTA, 1.2 NaH2PO4, 1.2 MgCl2 and 5.5 glucose, pH 7.4). The skull was divided in two halves and the MMA was carefully dissected out from the dura mater on the inner side of the skull under an operating microscope. Proximal segments of the MCA were dissected out from the brain, starting from the circle of Willis. Each vessel was cut into 1–2-mm-long circular segments and placed in an ice-cold Na+-Krebs buffer solution (in mM: 119 NaCl, 15 NaHCO3, 4.6 KCl, 1.5 CaCl2, 1.2 NaH2PO4, 1.2 MgCl2 and 5.5 glucose, pH 7.4), gassed with a 5/95% mixture of CO2/O2. To determine vessel tension, each segment was mounted on two metal wires 25 µm in diameter in a myograph (model 610M; Danish Myo Technology, Aarhus, Denmark). The buffer solution was continuously maintained at 37°C and aerated with a 5/95% mixture of CO2/O2 to maintain a stable pH of 7.4. The artery segments were allowed to equilibrate for approximately 30 minutes. The vessels were stretched to the internal circumference the vessel would have had if relaxed and exposed to a passive transmural pressure of 52 mmHg (7.0 kPa). This was done to achieve maximal active force development. Following a second 30-minute equilibration period, the vessels were constricted twice with 60 mM KCl in a modified buffer solution in which NaCl was substituted for KCl on an equimolar basis. The contraction amounted to 0.84 ± 0.09 mN for MMA (N = 37) and 1.04 ± 0.09 mN for MCA segments (N = 32).
Experimental design
To study the relaxant effect of PGE2 and butaprost, the MMA and MCA segments were precontracted with 10 µM 5-hydroxytryptamine (5-HT). In the MMA segments this resulted in a stable tension of 0.63 ± 0.09 mN or 73 ± 5% of the contraction induced by the 60 mM KCl buffer solution (N = 37). In the MCA segments it resulted in a stable tension of 0.62 ± 0.06 mN or 62 ± 4% of the contraction induced by the 60 mM KCl buffer solution (N = 32). Pilot experiments showed contraction properties of PGE2 at higher concentrations; therefore after achieving a stable precontraction, 1 µM of the thromboxane receptor (TP) antagonist, BAY-u3405, was added before the agonist was added in cumulative concentrations in a logarithmic fashion (PGE2 and butaprost: 10 nM–10 µM). Without these relaxants, the vessels were able to maintain a stable tone for the duration of the experiment. In experiments with antagonists, AH6809 (10 µM), BGC20-1531 (1 µM) or SQ22536 (30 µM) was added 15 minutes before preconstriction of the arteries. After preconstriction, another 15–20 minutes were allowed to pass prior to addition of PGE2 in increasing concentrations. The addition of antagonists per se did not affect the tension of the vessels. Out of three artery segments, one served as control, leaving the two other for experiments.
mRNA expression studies
Nine rats (350–400 g) were anesthetized with pentobarbital and perfused transcardially with 300 ml ice-cold Na+-Krebs buffer (in mM: NaCl 119, NaHCO3 15, KCl 4.6, CaCl2 1.5, NaH2PO4 1.2, MgCl2 1.2 and glucose 5.5) to wash out blood from the MMA, MCA, basilar artery (BA), trigeminal ganglion (TG) and trigeminus nucleus caudalis (TNC). The MMAs were carefully dissected out using a microscope, and immediately pooled in sterile Eppendorf tubes, containing an RNA stabilization solution (RNAlater; Ambion, Woodward, Austin, TX, USA). The arteries were placed at 4°C overnight and either further processed or stored at −80°C for later use. The RNA-stabilized arteries were disrupted and homogenized thoroughly with a rotor-stator homogenizer. Total RNA was purified from the arteries using an RNeasy Mini Kit (Qiagen, Hilden, Germany) with proteinase K digestion and on-column deoxyribonuclease (DNase) digestion steps included as specified by the manufacturer. After purification, yield and purity of the RNA were assessed spectrophotometrically by measuring the absorbance at 260 nm and by determining the ratio 260/280 nm, respectively. A total of three pooled fractions (total RNA from tissues of three rats per tube) were prepared for analysis. cDNA was synthesized from 1µg purified total RNA in a final reverse transcription PCR (RT-PCR) using the QuantiTect Rev. Transcription Kit (Qiagen, Hilden, Germany) as specified by the manufacturer’s instructions.
Detection of the molecular components relevant for rat PGE2 receptors and β-actin

Each set of primers was designed from the US National Center for Biotechnology Information (NCBI GenBank) nucleotide sequences.
Data and statistical analysis
GraphPad Prism® (GraphPad Software Inc., CA, USA) was used for all statistical analysis of all performed experiments and for the construction of the graphs. Statistical significance was assumed when p < .05.
Analysis of the in vivo studies was performed as previously described (31). In brief, they were based on measurements of two parameters: the changes in the diameter of the artery and changes in MABP. The vessel diameter was measured in arbitrary units. The MABP was given in mmHg. Dilatation of the vessels and changes in MABP were calculated as percentage change from the baseline, which was defined as the average of the 60 seconds preceding the administration of test substance. Vessel diameter was measured at the peak response, occurring one to two minutes after drug administration. All data are expressed as the mean ± standard error of the mean (SEM) of the percentage change from baseline values, with N indicating the number of experiments. The dose-response curves were analyzed to establish the maximum response (Emax ) and the negative logarithm of the dose (pED50) that leads to 50% of the maximal response. In case of topical administration, pEC50 was calculated as the concentration required, increasing the artery diameter by 50% of Emax . For inhibitor or antagonist experiments the ‘percentage inhibition’ was calculated as the percentage difference between the mean maximal inhibition compared to mean PGE2 response (300 ng kg−1). When comparing consecutive responses to either saline or PGE2, a one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test was used.
For the in vitro studies E max (maximum relaxant effect obtained with PGE2 or butaprost) and pEC50 (negative logarithm of the concentration of agonist that elicited half-maximum relaxation) were calculated from each individual concentration-response curve. Values are given as mean ± SEM; number of experiments = n; one MMA segment from each rat. The nonparametric, Mann-Whitney U-test was used to determine statistical significance between the two groups of data.
For the mRNA expression studies the final results were expressed as a target/reference ratio, normalized by the target/reference ratio from a calibrator. In these experiments a mix of kidney and lung calibrator was used (32,33). For data analysis the target/reference ratios in both samples and calibrator were determined in duplicates. All fluorescence data were processed with the LightCycler software from Roche (version 1.0). The paired t-test was used to determine the statistical difference between the two groups.
Drugs
Rat α-CGRP was obtained from PolyPeptide Laboratories (Strasbourg, France). Prostaglandin E2 (pKi 9.0 [EP4] and 8.2 [EP2] (1)), AH6809 (EP2 receptor antagonist; pKi 6.3 [EP2] (1)), BAY-u3405 (tTP antagonist; pKi 7.9 [TP] (34)), and butaprost (free acid) (EP2 receptor agonist; pKi 7.2 [EP2] and 4.8 [EP4] (1)) were from Cayman Chemicals Europe (Tallinn, Estonia). SQ22356 (adenylate cyclase inhibitor; IC50 20 µM (35)), was purchased from Sigma-Aldrich (Brøndby, Denmark). 5-HT was from Tocris Cookson Ltd., (Bristol, UK). BGC20-1531 (EP4 receptor antagonist; pEC50 7.6 [EP4] (28)) was a gift from BTG International Ltd., (London, UK). ONO-AE1-329 (EP4 receptor agonist; pKi 8.0 [EP4] and 5.7 [EP2] (26)) was a gift from ONO-pharmaceuticals (Osaka, Japan).
ONO-AE1-329 was dissolved in saline-added 0.3% ethanol and 0.1% Tween 80 to provide a stock solution of 2 mg ml−1. CGRP and 5-HT were dissolved in saline to provide a stock solution of 380 µg ml−1 and 2.2 mg ml−1, respectively. PGE2, AH6809, SQ22536, BGC20-1531, BAY-u3405 and butaprost were dissolved in dimethylsulfoxide (DMSO) to provide stock solutions of 4 mg ml−1, 1 mg ml−1, 2 mg ml−1, 8 mg ml−1, 417 µg ml−1 and 10 mg ml−1, respectively. For in vivo use all stock solutions were administered per se or further diluted in saline to their final concentrations just prior to experimentation. For in vitro use all stock solutions were further diluted in Na+-Krebs buffer to their final concentrations just prior to experimentation. Due to the use of DMSO as a solvent for some of the investigated drugs, a vehicle control series was made, as described in ‘Results'. The control experiments were performed the same way as the antagonist experiment series, as described above.
Results
In vivo effects of PGE2, butaprost and ONO-EA1-329 on rat middle meningeal artery
In the closed cranial window, PGE2 (1 ng kg−1–3000 ng kg−1), butaprost (1 µg kg−1–100 µg kg−1) and ONO-AE1-329 (1 ng kg−1–3000 ng kg−1) (all at half-log increments) caused dose-dependent dilatation of the rat MMA (Figure 2A). The mean pED50 values were 7.0 ± 0.31, 5.0 ± 0.17 and 7.4 ± 0.09, respectively. Only the highest doses administered with PGE2 and ONO-AE1-329 caused a significant decrease in MABP (Table 2). Administration of the highest dose of butaprost (100 µg kg−1) did not induce any changes in MABP (Table 2). Topical application of PGE2 (10 ng ml−1–30 µg ml−1) and butaprost (10 µg ml−1–300 µg ml−1) caused concentration-dependent dilatations of the MMA (Figure 2B). Mean pEC50 values were 6.1 ± 0.16 and 4.2 ± 0.10, respectively. Topical administration of these two compounds did not significantly alter MABP, even at the highest concentrations (data not shown).
(A) Effect of increasing doses of PGE2, butaprost and ONO-AE1-329 on rat middle meningeal (dural) artery diameter by i.c. infusion. *P < 0.05 (one-way ANOVA followed by Dunnett’s post hoc test) significantly different from vehicle infusion (n = 4–6). (B) Effect of increasing doses of PGE2 and butaprost on rat middle meningeal (dural) artery diameter by topical administration. *P < 0.05 (one-way ANOVA followed by Dunnett’s post hoc test) significantly different from topical vehicle dissolved in synthetic interstitial fluid (SIF) administration (n = 4). Agonist effects on MABP (%) administered via intra carotid infusion MABP = mean arterial blood pressure. nd = not determined.
p < .05, significantly different from MABP in the presence of vehicle.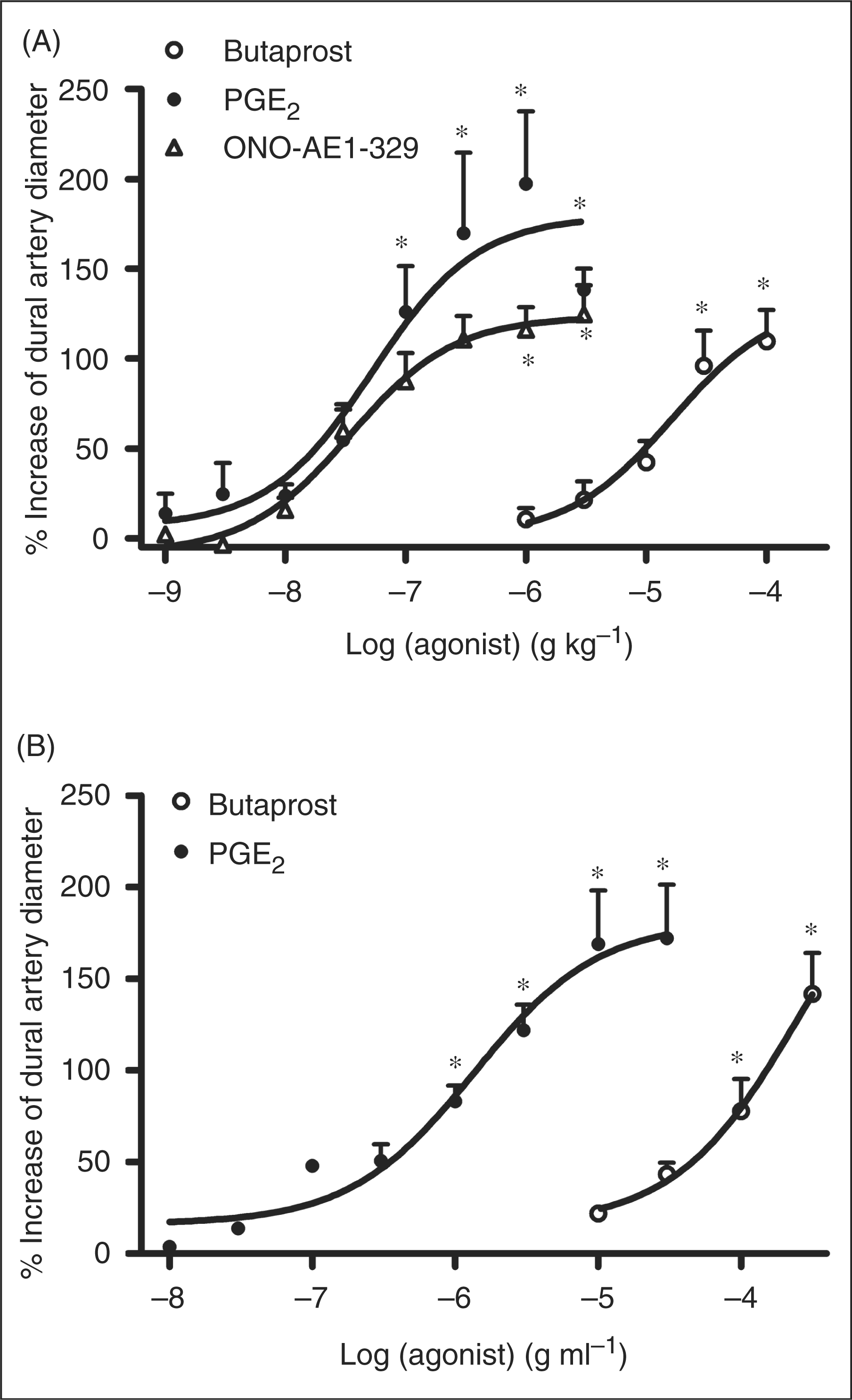

In vivo effects of PGE2 on rat middle cerebral artery
Administration of PGE2 i.c. (30 ng kg−1–3000 ng kg−1) to rat MCA did not induce a dilatory effect of rat cranial arteries, except at the highest dose (3000 ng kg−1), where a concomitant significant drop in MABP was observed (data not shown). Topical application of PGE2 (10 ng ml−1–30 µg ml−1) did not have any effect on MCA or MABP (data not shown).
Vehicle controls
As some antagonists used in this study were only soluble in DMSO, a control study was made to investigate the effect of DMSO on PGE2-induced dilatation on rat MMA. The results show no significant difference between the 300 ng kg−1 PGE2-induced dilatation and the dilatation induced by 300 ng kg−1 PGE2 following any dose (0–100%) of DMSO via i.c. administration (50 µl min−1) (Figure 3) (N = 5). DMSO caused a transient increase in dural artery diameter and a concurrent decrease in MABP (∼20%) from basal levels. Both these parameters reverted to basal levels within three minutes after which only PGE2 was administered.
The effect of increasing doses (0–100%) of DMSO on PGE2-induced response (300 ng kg−1) on rat middle meningeal (dural) artery diameter by i.c. infusion (n = 5). Each bar is expressed as the mean ± S.E.M. *P > 0.05 (One-way Anova followed by Dunnett’s post hoc test) the tested PGE2 responses after any DMSO treatment is not different from initial PGE2 responses.
In vivo effects of EP2 and EP4 receptor antagonists on PGE2-induced dilatation in rat middle meningeal artery
The PGE2-induced dilatations were characterized using the EP2 receptor antagonist AH6809 and the EP4 receptor antagonist BGC20-1531. In these experiments a submaximal dose of PGE2 of 300 ng kg−1 was used. The reproducibility of the PGE2 response was tested by repeating the dose three times prior to each experiment (Figure 1). The maximum dilatory responses were 109 ± 18%, 110 ± 19% and 113 ± 20% for each of the three dilatory peak responses (N = 3). AH6809 (120 µg kg−1), significantly blocked PGE2- (300 ng kg−1) induced dilatations by 40 ± 9% on MMA (Figure 4A). BGC20-1531 (100–3000 µg kg−1) significantly antagonized the PGE2-induced dilatation of MMA throughout the dose range tested (59 ± 6% at 3000 µg kg−1, Figure 4B).
Effect of the adenylate cyclase inhibitor, SQ22536, on PGE2-induced dilatations in rat middle meningeal artery
The adenylate cyclase inhibitor, SQ22536 (100 µg kg−1), inhibited the PGE2-induced dilatation more than 60% (Figure 4C).
Effects of EP2 and EP4 receptor antagonists on PGE2 (300 ng kg−1) induced dilatation on rat middle meningeal (dural) artery in vivo. (A) The EP2 receptor antagonist AH6809 (n = 4). (B) The EP4 receptor antagonist BGC20-1531 (n = 5). (C) The adenylate cyclase inhibitor, SQ22536 (n = 5). Each bar is expressed as the mean ± S.E.M. *P < 0.05 (one-way Anova followed by Dunnett’s post hoc test) significantly different from vehicle.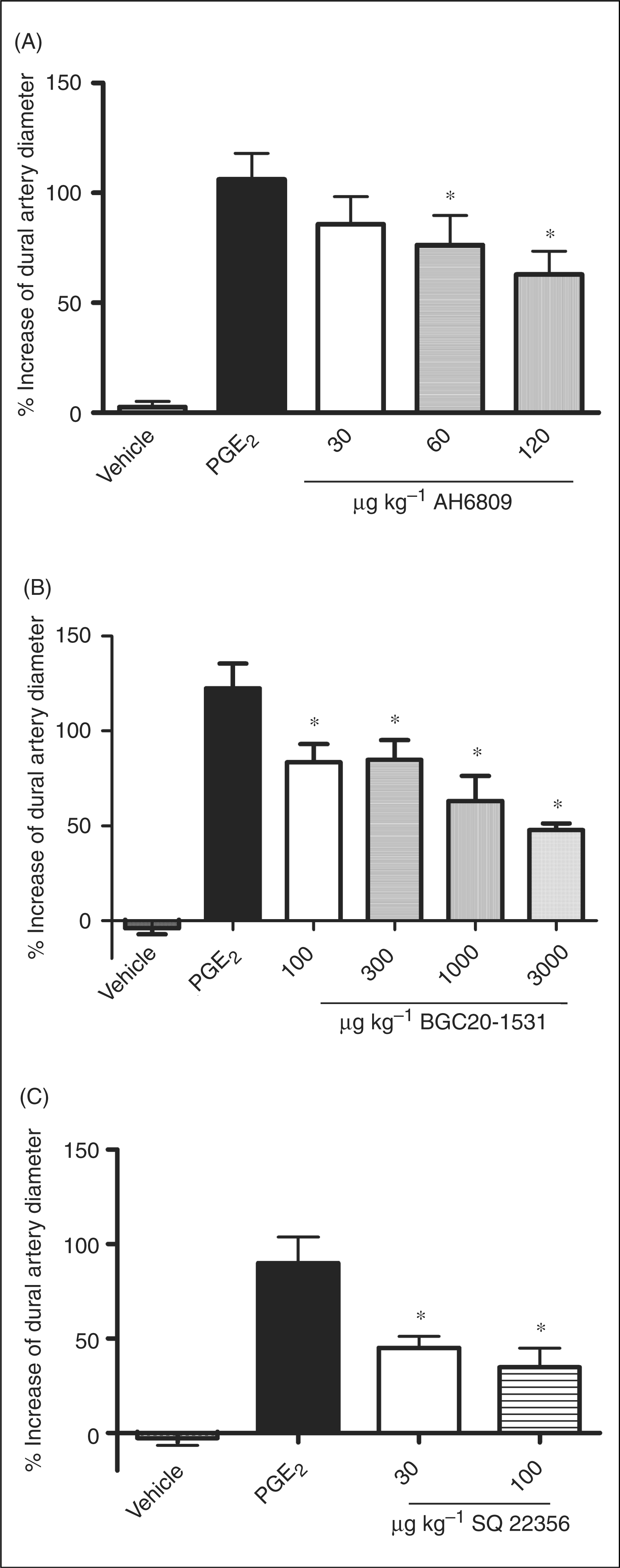
In vitro antagonism of the effects of PGE2 on isolated rat arteries
Isolated preparations of rat MMA and MCA were precontracted with 10 µM 5-HT producing a stable level of tension. Application of increasing concentrations of PGE2 (10 nM–10 µM) to the precontracted vessel segments resulted in relaxation of rat MMA (pEC50: 6.89 ± 0.18) and MCA (pEC50: 6.40 ± 0.09) (Figures 5A and 5B). Likewise, butaprost induced relaxation with similar potency on both rat MMA (Emax
: 60.76 ± 8.72% and pEC50: 5.99 ± 0.15) and MCA (Emax
: 42.04 ± 8.43% and pEC50: 6.54 ± 0.14). In rat MMA, precontracted with 10 µM 5-HT, the antagonists BGC20-1531, AH6809 and SQ22536 inhibited the PGE2-induced vasorelaxation (Figure 5A), with apparent pK
B values of ≈ 7 - 6 (Table 3). In these experiment series, the concentration-response curves to PGE2 significantly shifted rightward in presence of all tested compounds (Table 3).
Effect of (▴) AH6809 (10 μM) (n = 6), (○) SQ22536 (30 μM) (n = 5) and (□) BGC20-1531 (1 μM) (n = 4) on (•) PGE2-induced vasorelaxations in in vitro isolated rat middle meningeal artery (MMA) (A) and in isolated rat middle cerebral artery (MCA) (B). The relaxation of each segment was calculated as a percentage of the pre-contraction induced by 10 μM 5-HT. Each point represents mean values ± S.E.M.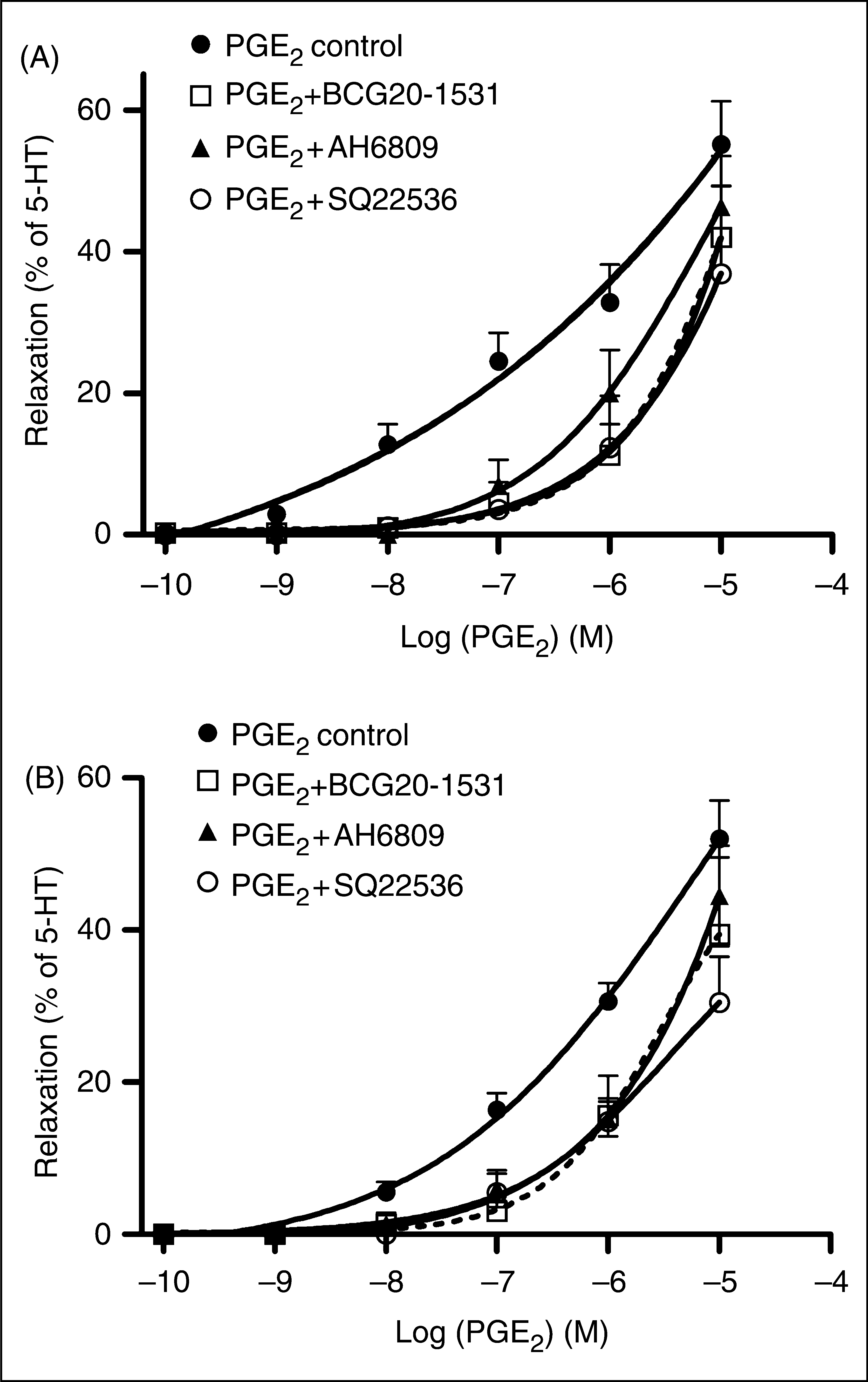
Effects of antagonist/inhibitor on relaxations to PGE2 in rat MMAs a

MMAs = middle meningeal arteries. MCAs = middle cerebral arteries.
Antagonist/inhibitor was incubated for 0.5 h, subsequently segments were preconstricted with 10 µM 5-HT. After attaining a stable preconstruction, PGE2 was added in concentrations increasing in half-log steps. Emax was expressed as % of the response to 10 µM 5-HT. All data are means ± S.E.M (N).
p < .05, significantly different from mean PGE2 pEC50.
Reverse transcription PCR and quantitative real time PCR analysis
Under identical RT-PCR conditions, mRNA expressions for all PGE2 receptors (EP1, EP2, EP3 and EP4) were verified in MMA, MCA, BA, TG and TNC (Figure 6). All PCR products were also present in positive control (kidney and lung mix) (32,33). All products were extracted and sequenced. We aligned the sequenced PCR products with the existing target sequence information from NCBI GenBank, which resulted in 99–100% nucleotide identity to all primer-generated EP receptor products.
mRNA expression of EP1-EP4 receptors in rat cranial arteries and neuronal tissue. RT-PCR products separated through an agarose gel in order to study mRNA expression in middle meningeal artery (MMA), middle cerebral artery (MCA), basilar artery (BA), trigeminal ganglion (TG) and trigeminus nucleus caudalis (TNC). Each primer pair tested generated only one product. Product sizes for all EP1-EP4 receptors were equal to the expected sizes (Table 1) estimated from rat target mRNA sequences (n = 3).
Quantification is usually measured by normalizing the detection threshold of specific primer pairs to detection threshold cycles of β-actin primers. However, EP2 and EP4 mRNA products were only detected at very late cycling stages. Hence, we utilized the product doubling function of the Relative Quantification (Roche) software in order to determine the quantification of the EP2 and EP4 mRNA products. From these results, EP2 and EP4 tended to be expressed in MMA = MCA>TG>TNC>BA (Figures 7A and 7B).
Quantitative mRNA expressions of (A) EP2 and (B) EP4 receptors in rat middle meningeal artery (MMA), middle cerebral artery (MCA), basilar artery (BA), trigeminal ganglion (TG) and trigeminus nucleus caudalis (TNC). Both receptors seem predominantly expressed in MMA, MCA over BA and TG over TNC. *P < 0.05, significantly different from MMA; #P < 0.05, significantly different from MCA. One-way ANOVA with Bonferroni Correction (n = 3).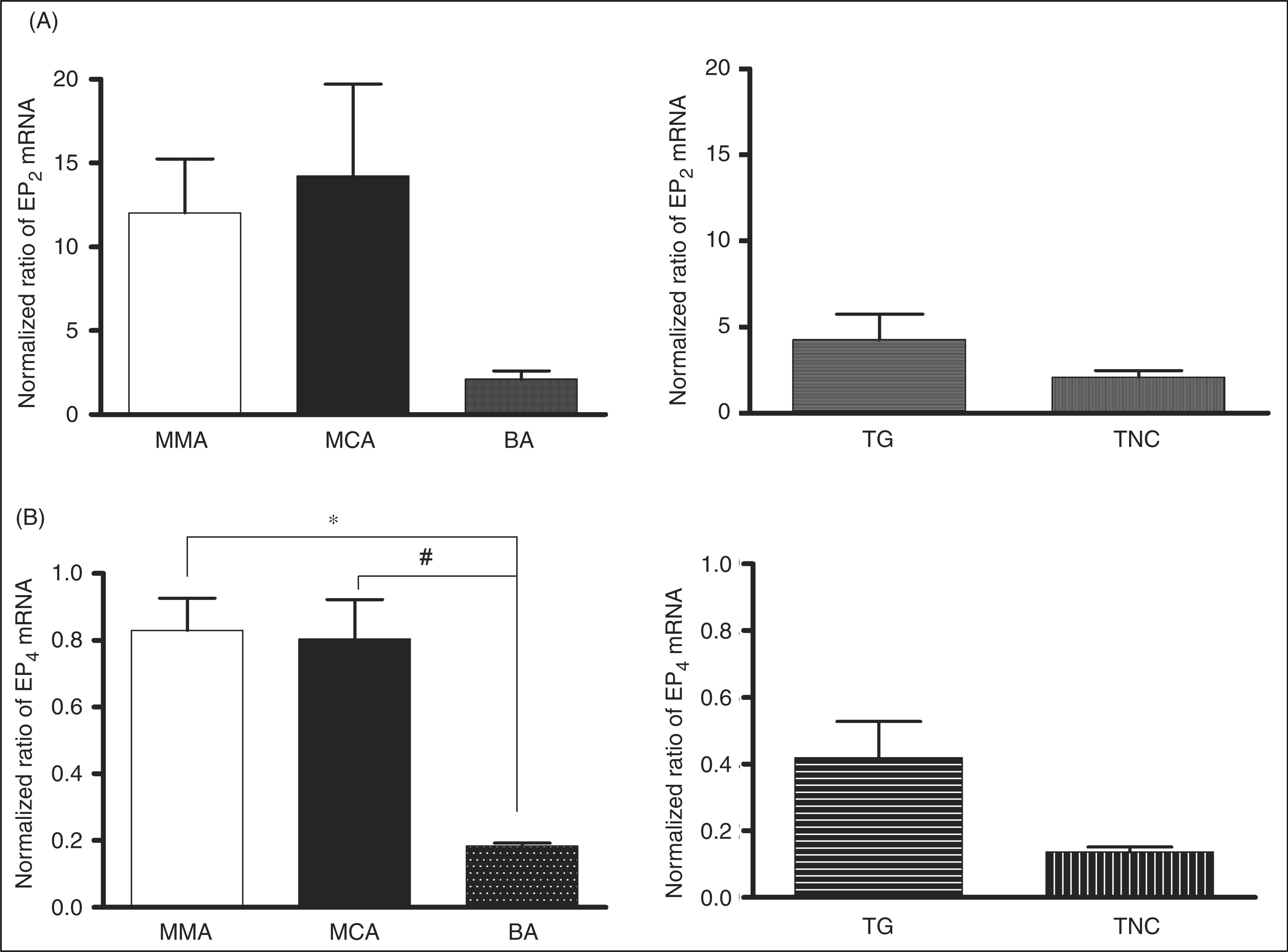
Discussion
This is the first study demonstrating and characterizing the vasodilatory role of PGE2 in rat MMA and MCA using specific agonists and antagonists of the EP2 and EP4 receptors. Additionally, we show EP1-EP4 mRNA transcripts and quantify EP2 and EP4 receptors in the trigeminal vascular system.
Intracarotid as well as topical administration of PGE2 induced dose/concentration-dependent dilatation of MMA in the rat closed cranial window model. These results indicate that receptors for PGE2 are present in the rat MMA, and that PGE2 can penetrate the thinned skull to act on the underlying MMA thereby mimicking a physiological condition, in which perivascular nerves could release prostaglandins around the smooth muscle cells of cranial vessels. To our knowledge, no study of PGE2 dose-dependent dilatation of MMA has been published before for any species. When investigating the MCA response to increasing concentrations of PGE2, no response was seen by topical (at any dose) administration. Therefore, our studies indicate that PGE2 does not pass the blood-brain barrier in rats. Also, dilatation was not detectable with i.c. administration of PGE2, except at high doses. These doses caused dilatation of MCA, probably due to autoregulation induced by the concurrent decrease in blood pressure (22,23). The lack of dilatation could also be due to low or no presence of EP2 and EP4 receptors on the endothelial cells lining this vessel. Previously, studies in the cat open cranial window model has shown that topically applied PGE2, 0.0353–3.525 µg ml−1, induces dilatation of large and small pial arterioles (36). Likewise, an in vivo piglet open window model showed topically applied PGE2, 1–100 ng ml−1, as a vasodilator of pial arteries (37). From the present results and the fact that similar concentrations have been used in previous work (36,37), it seems probable that topically applied PGE2 might not reach cerebral arteries in our model, likely due to the arachnoid membrane posing an impermeable barrier for this prostanoid.
The dilatory response to PGE2 is usually mediated via EP2 and EP4 receptors in the vascular beds, but this has never before been demonstrated in rat cranial arteries. Therefore, we studied the EP2 receptor agonist, butaprost, and the EP4 receptor agonist, ONO-AE1-329. Our results show that ONO-AE1-329 is equipotent to PGE2 and more than two log units more potent than butaprost (pED50: 7.4 vs. 5.0), although the reported affinity of butaprost for the EP2 receptor is 7.2 (1) and the affinity of ONO-AE1-329 at the EP4 receptor is 8.0 (26) at rat and mouse receptors. Recently, ONO-AE1-329 has been reported to have an effect similar to that of PGE2 in relaxing human pulmonary veins (pEC50: 7.80 ± 0.09 vs. 7.22 ± 0.20) (38), thus supporting our findings. Together, these results suggest that although EP2 receptors are present, they play a lesser role in relaxation than EP4 receptors in the rat cranial arteries.
The EP2 receptor antagonist, AH6809, at its highest dose attenuated the PGE2 diameter response by 40%, and the novel EP4 receptor antagonist, BGC20-1531, inhibited the PGE2-induced dilatation by 60%. A recent study (28) showed that BGC20-1531 antagonizes PGE2-induced dilatation in the canine vascular bed with an ID50 of ∼5 mg kg−1 administered i.v. Similarly, in our hands 3mg kg−1 of the antagonist, which was administered i.c., also inhibited more than 50% of the PGE2 response, though the differences in route of administration and species should be noted between the experimental setups. In conclusion, our findings show that both the EP2 and EP4 receptors play a role in PGE2-mediated dilatation of rat cranial arteries. As PGE2 infusion in humans has already been shown to provoke headaches in humans, it is highly likely that EP2 and EP4 receptors might be the key transducers of PGE2 provoked headaches. Furthermore, PGE2 levels are reported to be increased in the blood during migraine attacks (12,15), which could cause a dilatation of the cerebral and meningeal vasculature, contributing to the migraine pain experience.
EP2 and EP4 receptors signal primarily through the second messenger cAMP. We also confirm involvement of this second messenger system in the dural arteries, as the adenylate cyclase inhibitor, SQ22536, was effective in blocking the PGE2-induced dilatation.
Importantly, in vitro rat MMA and MCA were also relaxed by PGE2, in the presence of the TP receptor antagonist BAY-u3405. Although the concentration-response curves in this study did not reach a plateau in the concentration range tested, the obtained values are comparable to previously published values in human MCA (pEC 50: 8.0 ± 0.1) (39) and recently in canine MMA (mean pEC 50: 7.6 ± 0.2) (28). To characterize these involved receptors in vitro, two antagonists and one inhibitor were used. BGC20-1531 gave an apparent pK B value of 7.34 ± 0.15 in rat MCA and 7.01 ± 0.54 in rat MMA. A similar result was seen in human MCA and MMA; in fact, very similar pK B values were seen between HEK-293 EBNA cells stably transfected with EP4 receptor, human cerebral and meningeal arteries as well as dog MMA (28). It has been suggested that EP4 receptors might be a therapeutic target in the treatment of pain due to the ability to attenuate inflammation-induced thermal and mechanical behavioural hypersensitivity after delivering EP4 receptor short hairpin RNA to mice intrathecally (40). Furthermore, inhibition of this receptor exhibits antihyperalgesic effects in animal models of acute and chronic inflammation pain (41). Likewise, the vasodilatory receptor EP2 was antagonized by AH6809. Using the adenylate cyclase inhibitor, the maximum inhibition we observed in the in vitro experiments was seen suggesting an antagonism of both EP2 and EP4 receptors via cAMP inhibition. Thus, our results in vitro also support the existing understanding of PGE2 signaling via EP2 and EP4 receptors.
In current expression studies, mRNA of EP1-EP4 receptors was present in all the investigated rat arteries and neuronal tissues. Accordingly, these data are in agreement with mRNA expression in human MCA (39). The quantitative expression studies of the craniovascular arteries indicate that EP2 and EP4 seem mainly located on the MMA and MCA, as compared to the BA. Logically, resistance vessels would have a higher distribution of dilatory receptors than conducting vessels. There is a tendency toward more EP2 and EP4 receptor mRNA expression in TG compared to TNC, although this observation is not significant. An extensive report on EP2 and EP4 receptor distribution in the rat brain has been published (33) but did not deal with the craniovascular system. Previous work (21) shows that mRNA expression in whole excised trigeminal ganglia expresses all four types of EP receptors. Therefore, to our knowledge, this is the first study to show quantitative mRNA expression of EP2 and EP4 receptors in the rat cranial arteries, TG and TNC. The fact that the receptors very likely are present in these tissues indicates that the trigeminal nerve can be activated by PGE2 directly. This is supported by studies showing that PGE2 can release CGRP from rat trigeminal ganglion neuronal cultures (21), and provides an additional link to the relevance of PGE2 in migraine.
In conclusion, we have shown that PGE2 can dilate rat meningeal arteries in vivo and relax both isolated rat MMA and MCA in vitro. mRNA of all EP receptors was present in cranial arteries, TG and TNC structures comprising the trigeminal vascular system. Quantitatively, EP2 and EP4 receptor mRNA seemed to be predominant in the meningeal arteries. Both EP2 and EP4 receptor antagonists attenuated PGE2-induced responses—a response most likely mediated via cAMP signaling. Furthermore, our data point toward functional predominance of EP4 receptors in these preclinical models. Thus, the data indicate that EP2 and particularly EP4 receptor antagonists may have therapeutic utility in the treatment of migraine pain.
Footnotes
Acknowledgments
The study was supported by the Lundbeck Foundation as part of the Lundbeck Foundation Center for Neurovascular Signaling (LUCENS), by the Augustinus Foundation and by the Danish Medical Council and the Danish agency for Science Technology and Innovation (No-271-07-0773). We would like to thank both BTG International Ltd. for their kind gift of BGC20-1531 and likewise ONO-pharmaceuticals for ONO-AE1-329. Special thanks to Dr. K. Maubach for useful comments on the manuscript.