
Abstract
Migraine is associated with stroke-like episodes in mitochondrial encephalomyopathy, lactic acidosis, stroke-like syndrome (MELAS). Moreover, abnormalities of oxidative phosphorylation are also reported in migraine. We studied two maternal lineages with MELAS and chronic progressive external ophthalmoplegia (CPEO) affected probands carrying the 3243 A>G tRNALeu (MELAS) mutation, remarkable for a high frequency of subjects suffering only migraine. Thus, migraine could be a monosymptomatic expression of the MELAS mutation. We assessed the 3243 A>G tRNALeu mutational load in skeletal muscle and other somatic tissues from the migraine-only subjects, as well as lactic acid levels after exercise. All migraine-only subjects did not carry the MELAS mutation. Muscle biopsy showed mild mitochondrial abnormalities in the non-mutant, migraine-only subjects and, occasionally, abnormal lactic acid. Clear features of mitochondrial myopathy and pathological lactic acid characterised the subjects carrying the MELAS mutation. Our study demonstrates that migraine-only subjects lacked the MELAS mutation, but still had a possible mtDNA-associated genetic predisposition, being maternally related and having some evidence of impaired mitochondrial oxidative phosphorylation.
Introduction
Migraine features have been recognised as characteristically associated with the stroke-like events in mitochondrial encephalomyopathy, lactic acidosis, stroke-like syndrome (MELAS) due to the most common mitochondrial DNA (mtDNA) point mutation at position 3243 A>G tRNALeu (1,2). Migraine has also been reported as a frequent, non-specific feature of MELAS, similar to sensorineural deafness, diabetes or short stature, which may characterize oligosymptomatic cases carrying a low mutational load (3). Furthermore, abnormalities of mitochondrial oxidative phosphorylation have been documented in the common forms of migraine, with or without aura, using phosphorus magnetic resonance spectroscopy ([31P]-MRS) or assessing respiratory enzyme activities in subject-derived tissues (4–9). Together, these findings support the hypothesis of defective oxidative phosphorylation in the pathogenesis of migraine (10–12).
The occurrence of migraine has been reported in other primary mitochondrial disorders such as in families with Leber’s hereditary optic neuropathy (LHON), or in a subject carrying a single mtDNA deletion and suffering migraine strokes (13–15). The common MELAS mutation was also found in a subject suffering cluster headache, but this association was not confirmed in a subsequent study on a larger case series (16,17). Indeed, systematic studies screening for mtDNA mutations in a series of migraine-only subjects, all failed to recognize an association (18–22). However, all these studies were substantially flawed by the fact that the mutant mtDNA was looked for in blood-derived cells, where severe mtDNA mutations, such as the common MELAS mutation, become undetectable in an age-dependent fashion due to selection (23). Other studies checked for the possible association of mtDNA polymorphic variants or haplogroups in migraine subjects, claiming that specific variants of mtDNA may represent a predisposing factor (24).
In the current study, we undertook a different strategy, investigating the occurrence of the common mtDNA 3243 A>G tRNALeu MELAS mutation in skeletal muscle from migraine-only subjects belonging to two unrelated maternal lineages with chronic progressive external ophthalmoplegia (CPEO) or MELAS-affected subjects carrying this mutation.
Subjects and methods
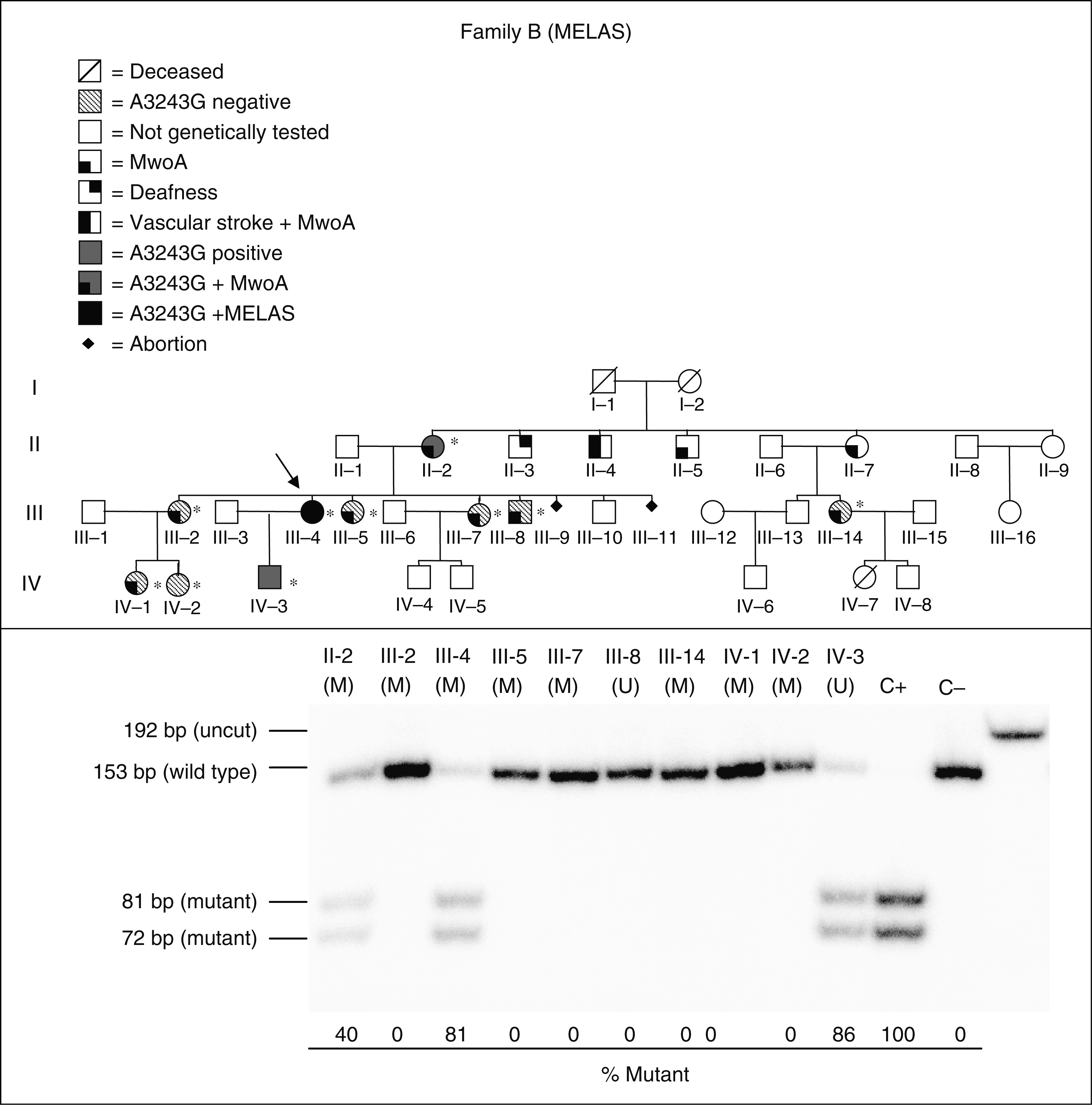
We studied two unrelated Italian families carrying the mtDNA 3243 A>G tRNALeu MELAS mutation. The proband in family A (arrow in Figure 1) was affected with CPEO, whereas the proband from family B (arrow in Figure 2) had full blown MELAS. In both maternal lineages, a high recurrence of migraine-only subjects was present (Table 1), as evaluated by clinical interview in all maternally related subjects. The headache diagnosis was based on the International Headache Society (IHS) criteria (25). We report the detailed clinical description only for the subjects who were positive for the 3243 A>G tRNALeu MELAS mutation. The clinical features of the other subjects belonging to both maternal lineages, who underwent further investigations, including muscle biopsy, are summarised in Table 1. A few other cases underwent only genetic analysis (subjects II-6 and IV-1 in family A (Figure 1) and IV-2 and IV-3 in family B (Figure 2)).
Pedigree and genetic analysis of family A. Upper panel: pedigree of family A. The arrow indicates the proband. Asterisks indicate subjects who underwent genetic evaluation. Solid grey indicates A3243G positive subjects, whereas line pattern indicates A3243G negative subjects. Lower panel: genetic evaluation of MELAS mutation load in skeletal muscle and urinary epithelium. Pedigree and genetic analysis of family B. Upper panel: pedigree of family B. The arrow indicates the proband. Asterisks indicate subjects who underwent genetic evaluation. Solid grey indicates A3243G positive subjects, whereas line pattern indicates A3243G negative subjects. Lower panel: genetic evaluation of MELAS mutation load in skeletal muscle and urinary epithelium.
Family A (CPEO)
Subject II-2 (proband)
This 61-year-old woman had suffered severe migraine without aura (MwoA) since the age of 26 years, with a frequency of 2–3 attacks per month. She also presented bilateral ptosis and CPEO since the age of 36 years. At 50 years of age, she also complained of progressively worsening proximal muscle hyposthenia and restless leg syndrome (RLS). Neurological examination showed bilateral ptosis (left > right), bilateral ophthalmoplegia, mild weakness of temporal, orbicular, neck and shoulder girdle muscles. EEG and brain CT scan were normal.
Subject III-2
This 36-year-old woman has suffered moderate MwoA 4–6 times per month since she was 22 years old. She also had nocturnal muscle cramps. Neurological examination was unremarkable except for pes cavus and scoliosis. EEG showed left frontotemporal theta activity. Visual and brainstem evoked potentials were normal, as well as brain CT scan.
Subject IV-1
The 15-year-old son of subject III-2 did not undergo clinical investigations. He did not complain of any neurological symptom, but he has been treated with growth hormone because of his short stature.
Family B (MELAS)
Subject III-4 (proband)
This 33-year-old woman had her first stroke-like episode preceded by a migraine-like headache with vomiting and loss of vision when she was 31 years old. Her symptoms progressed over the next 2 years with recurrence of multiple stroke-like episodes, frequently associated with migraine-like attacks and seizures. She also developed sensorineural deafness. Neurological examination showed severe cognitive impairment, right homonymous lateral hemianopsia, horizontal and vertical gaze-evoked nystagmus, hypotonia, right hemiparesis and right vibratory sensory loss. Interictal EEG showed left theta activity more evident on the occipital region. Brain MRI revealed a large area of increased T2 signal involving the left occipital lobe and the left posterior temporal-parietal region.
Subject II-2
This 55-year-old proband’s mother had suffered MwoA once a month since she was 25 years old. She reported moderate unilateral (right) migraine attacks. Neurological examination showed gaze-evoked nystagmus, intentional tremor more evident at eyes closed. EEG revealed left theta activity. Brain MRI was normal.
Subject IV-3
The 8-year-old son of the proband did not undergo clinical investigations and he did not complain of any neurological symptoms.
Genetic analysis
Total DNA was extracted from blood cells, urinary epithelium and skeletal muscle by the standard phenol/chloroform method. Detection of the 3243 A >G tRNALeu mutation and evaluation of mutational load was determined by restriction fragment length polymorphism (RFLP) analysis after PCR amplification. A 192 bp mtDNA amplicon encompassing the 3243 A>G tRNALeu mutation was obtained using the primer pairs: forward (nt3162–3184) and backward (nt3353–3337). After 25 cycles (94°C for 1 min, 57°C for 1 min, 72°C for 1 min), the amplified products underwent a last hot cycle in the presence of [α-32P]-dATP with a final extension at 72°C for 10 min. RFLP analysis was performed after overnight digestion with HaeIII by fragment separation on a 12% non-denaturing polyacrylamide gel. The wild-type pattern consisted of two fragments (153 bp and 39 bp) and the mutant of three fragments (81, 72, and 39 bp). The heteroplasmic condition was characterised by the co-occurrence of four bands (153, 81, 72 and 39 bp) and mutant/wild-type ratio was quantified by scanning the gel in a PhosphorImager (Bio-Rad, Model GS-363).
Muscle biopsy
Needle muscle biopsies of the quadriceps (vastus lateralis) were performed under local anaesthesia and after having obtained informed consent. Muscle specimens were frozen in cooled isopentane, stored in liquid nitrogen for standard histological and histo-enzymatic stains including Gomori modified trichrome, cytochrome-c oxidase (COX) and succinate dehydrogenase (SDH) activities according to standard protocols.
Serum lactic acid after standardised exercise
Serum lactate was evaluated at rest and after standardised exercise in all subjects, as previously reported (26).
Results
Genetic analysis
The initial screening for the MELAS mutation in blood cells showed variable amounts of mutant mtDNA in three subjects from family A (II-2, 5%; III-2, 11.5%; IV-1, 27%) and in two subjects from family B (III-4, 6%; IV-3, 22%). We have been able to collect eight muscle biopsies for each family. The MELAS mutation was detected in skeletal muscle in only two subjects (II-2, 39% mutant load and III-2, 44% mutant load) from family A (Figure 1, lower panel), and in two subjects (II-2, 40% mutant load and III-4, 81% mutant load) from family B (Figure 2, lower panel).
We collected urinary epithelium cells in those subjects who did not have muscle biopsy (I-2, II-6 and IV-1 from family A and III-8 and IV-3 from family B).
Screening for MELAS mutation in these subjects revealed the presence of 75% mutant load in subject IV-1 from family A (Figure 1, lower panel) and 86% mutant load in subject IV-3 from family B (Figure 2, lower panel).
Overall, only three subjects from each maternal lineage carried the MELAS mutation at variable loads and the large majority of subjects suffering from MwoA (5 of /7 in family A and 6 of 8 in family B) were negative for MELAS mutation.
Main clinical, radiological and EEG features of patients not carrying the MELAS mutation

Serum lactic acid after standardised exercise
Lactic acid and muscle biopsy findings
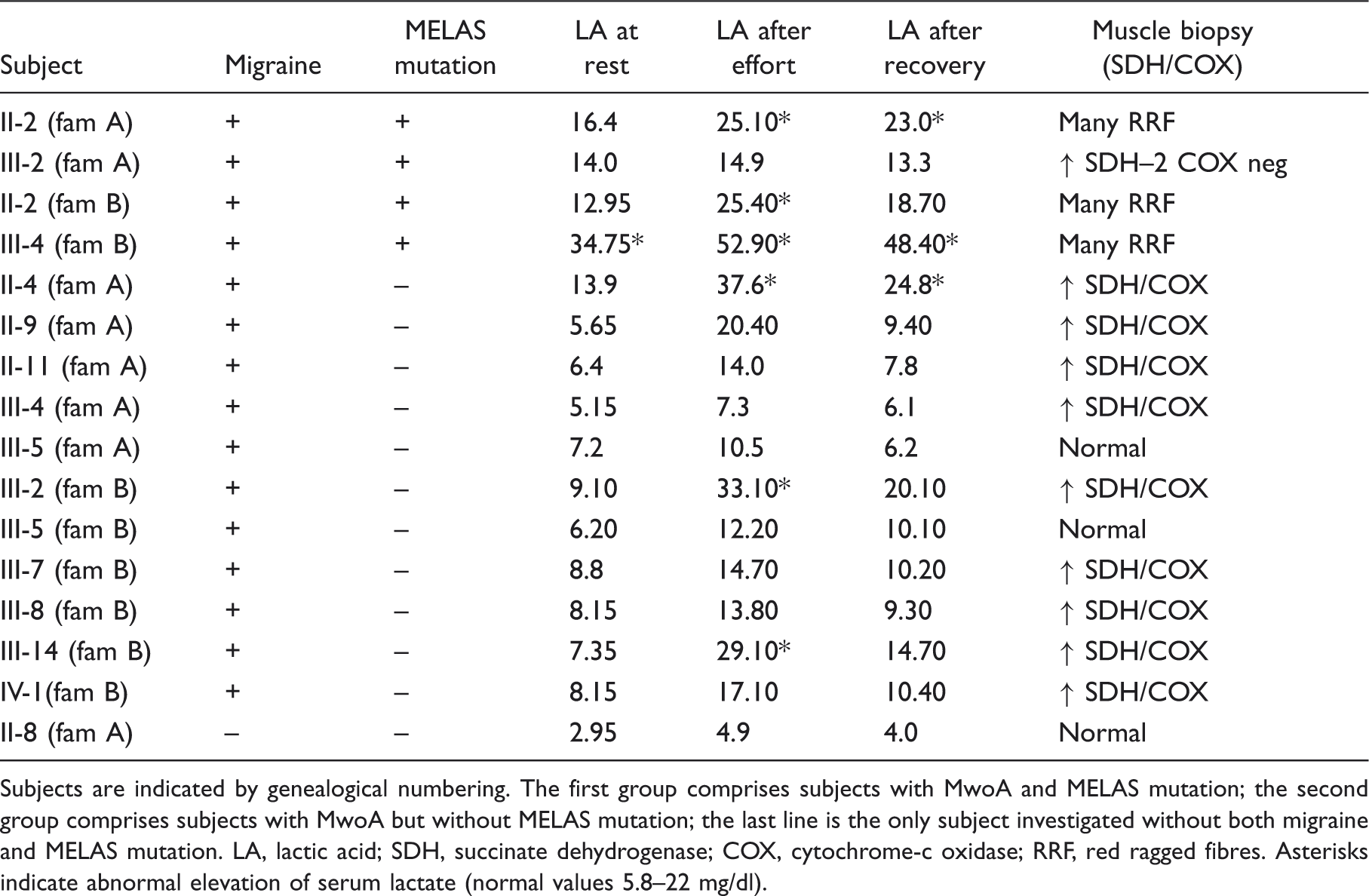
Subjects are indicated by genealogical numbering. The first group comprises subjects with MwoA and MELAS mutation; the second group comprises subjects with MwoA but without MELAS mutation; the last line is the only subject investigated without both migraine and MELAS mutation. LA, lactic acid; SDH, succinate dehydrogenase; COX, cytochrome-c oxidase; RRF, red ragged fibres. Asterisks indicate abnormal elevation of serum lactate (normal values 5.8–22 mg/dl).
Muscle biopsy
The histo-enzymatic results of muscle biopsies are also reported in Table 2. All subjects carrying the MELAS mutation (II-2 and III-2 from family A; II-2 and III-4 from family B) had some abnormalities of oxidative enzymes. Three out of four had classical and wide-spread ragged-red fibres (RRF), both COX positive and COX negative, and clear evidence of mitochondrial myopathy (II-2 from family A; II-2 and III-4 from family B; Figure 3A–D). Subject III-2 from family A showed rare hypotrophic fibres with increased subsarcolemmal SDH reaction and two COX negative fibres. All subjects without MELAS mutation but with MwoA, except two cases (III-5 from family A and III-5 from family B), showed mild abnormalities of oxidative enzymes distribution (SDH and COX), such as a partial subsarcolemmal enhancement in some fibres and occasional COX negative fibres (Figure 3B,C,E,F).
Muscle biopsies of subjects with and without 3243 A>G tRNALeu MELAS mutation. COX (upper panels) and SDH (lower panels) stain of muscle biopsies. (A,D) Muscle biopsy of subjects III-4 from family B. This was the proband affected with MELAS and carrying 81% mutational load. Asterisks indicate SDH/COX hyperintense fibres. (B,E) Muscle biopsy of subject III-4 from family A. (C,F) Muscle biopsy of subject III-2 from family B. Both subjects were negative for MELAS mutation and suffered only MwoA. Arrows indicate slight subsarcolemmal increase of SDH and COX staining suggestive of a modest mitochondrial proliferation.
These results indicate a frequent occurrence of mild muscle abnormalities of oxidative enzymes in the subjects with MwoA not carrying the MELAS mutation.
Discussion
Our investigation of two maternal lineages with CPEO or MELAS subjects carrying the 3243 A>G tRNALeu mutation and with a high recurrence of migraine-only subjects led to the result that most of the latter did not carry the mutation. At difference with the previous studies on migraine case series searching for the 3243 A>G tRNALeu mutation in blood cells (18–22), the current study assessed for the first time the mutational load in skeletal muscle or urinary epithelium, both tissues known for being more reliable to detect the MELAS mutation (23,27). Although very unlikely, we cannot completely exclude the presence of the MELAS mutation in other target tissues, such as brain capillary endothelium, not available for genetic investigations. Furthermore, this study investigated two pedigrees with migraine-only subjects, known for having probands carrying the MELAS mutation and not unselected series of migraine subjects.
In this study, we also had the rare opportunity to show muscle histology at biopsy in migraine subjects not carrying the MELAS mutation. Previous MR-spectroscopy investigations in migraine subjects with and without aura documented an abnormal oxidative phosphorylation in skeletal muscle (6,7). Our current data confirm these seminal observations by an unrelated approach. In fact, evidence of moderate increase in mitochondrial biogenesis was present at histo-enzymatic SDH/COX stains in most of the muscle biopsies from our migraine-only subjects (9 out of 11). Furthermore, serum lactic acid after effort was abnormally elevated in three out of 11 migraine-only subjects, as previously noted (28). Presence of moderate subsarcolemmal increase at SDH/COX stains may be frequently overlooked or considered normal, occurring also in normal subjects under different conditions, such as muscle training (29). Thus, a systematic investigation of this issue in a larger migraine-subject population is warranted by using the combination of different techniques such as MR-spectroscopy, evaluation of lactic acid after exercise, muscle histology, biochemical assays of respiratory complexes and gene expression studies. The combination of these investigations may define a subgroup of migraine patients with documented oxidative phosphorylation deficiency that could be suitable for more targeted therapies. Non-specific neurological abnormalities, such as theta EEG activity, nystagmus, and unsteadiness during migraine attacks observed in some of our subjects, are frequently reported in patients suffering migraine (30,31). Furthermore, we noted the coincidental occurrence of RLS in some individuals from family A, as all except one (II-8) also suffered MwoA. An association of migraine and RLS has been recently reported (32).
This study provides new clues on the clinical expression of the 3243 A>G tRNALeu mutation. Based on the current results, typical MwoA may not be a specific monosymptomatic feature of MELAS, as previously suggested (3). In fact, in our two families, migraine apparently segregated along the maternal line without necessarily co-segregating with the MELAS mutation. Thus, a different genetic predisposition seems to underlie migraine in these subjects unrelated to the MELAS mutation (12,22). The strict maternal relationship among the MwoA subjects not carrying the MELAS mutation still suggests an involvement of mtDNA, as previously reported (24,33). An attractive hypothesis is that the specific mtDNA haplotype of these two families may play a role, possibly in conjunction with nuclear genes, according to the current concept of a polygenetic determination for migraine predisposition. This issue can be resolved only by the complete mtDNA sequence of these two families searching for private variants. To test this hypothesis further, analysis of mtDNA haplogroup distribution in large cohorts of migraine subjects is warranted. To this end, a recent report provides evidence that mtDNA haplogroup may influence the therapeutic response to riboflavin in migraine patients (34).
The relationship between migraine features in MELAS and the occurrence of stroke-like episodes has long been discussed. An interesting hypothesis has been recently put forward to explain the selective distribution of ischaemic lesions in the posterior cortex in stroke-like episodes, possibly resulting from the combination of the vascular mitochondrial dysfunction in MELAS subjects (35,36) with the cortical spreading depression that has been documented in the migraine aura (37,38). Thus, migraine could represent an over-imposed precipitating factor for strokes in MELAS subjects, while being not necessarily related to the MELAS mutation. In this scenario, subjects carrying even high loads of MELAS mutation in the brain may not suffer life-long strokes if migraine-free, as frequently seen in some MELAS pedigrees (3). Indeed, even subjects suffering from migraine with and without aura, but not carrying the MELAS mutation, are at increased risk for subclinical ischaemic brain lesions (39). Again, a recent study suggested that mtDNA remains a relevant genetic factor also in these cases, in particular migraine subjects with occipital strokes, which were associated with the mtDNA haplogroup U5 in a Finnish population (40). Based on these observations, a preventive and acute migraine treatment in subjects carrying the MELAS mutation might be warranted to reduce the risk of stroke-like episodes, possibly triggered by a migraine attack.
Conclusions
This study has indicated that that typical MwoA may be an unrelated genetic trait driving the clinical expression of stroke-like episodes in subjects carrying the MELAS mutation, as recently hypothesised. Nonetheless, we show that oxidative phosphorylation dysfunction may still underlie migraine in the absence of the MELAS mutation, possibly depending on specific mtDNA haplotypes. Furthermore, these results confirm, and are compatible with, the polygenetic nature of migraine liability and with the multiple pathophysiological mechanisms involved in migraine pathogenesis.
Footnotes
Acknowledgements
The authors are indebted to the patients and their families for their invaluable collaboration. The financial support of Bologna University grant (RFO-ex 60%) to PC, PM and VC is also gratefully acknowledged.