
Abstract
Opioids remain the major drug class for the treatment of acute, chronic and cancer pain, but have major harmful effects such as dependence and opioid-induced ventilatory impairment. Although no new typical opioids have come onto the market in the past almost 50 years, a plethora of new innovative formulations has been developed to meet the clinical need. This review is intended to shed light on new understanding of the molecular pharmacology of opioids, which has arisen largely due to the genomic revolution, and what new drugs may become available in the coming years. Atypical opioids have and are being developed which not only target the mu opioid receptor but other targets in the pain pathway. Biased mu agonists have been developed but remain ‘unbiased’ clinically. The contribution of drugs targeting non-mu opioid receptors either alone or as heterodimers shows potential promise but remains understudied. That gene splice variants of the mu opioid receptor produce multiple receptor isoforms in different brain regions, and may change with pain chronicity and phenotype, presents new challenges but also opportunities for precision pain medicine. Finally, that opioids also have pro-inflammatory effects not aligned with mu opioid receptor binding affinity implicates a fresh understanding of their role in chronic pain, whether cancer or non-cancer. Hopefully, a new understanding of opioid analgesic drug action may lead to new drug development and better precision medicine in acute and chronic pain relief with less patient harm.
Introduction
Opioids remain the oldest group of drugs in the therapeutic armamentarium for pain relief. Over 4000 years ago Assyrian clay tablets contained instructions for collecting the juice of the poppy plant Papaver somniferum to treat pain, diarrhoea and insomnia, but with the harmful effect of addiction in some but not all users. Following Friedrich Sërturner’s isolation of morphine in 1817 and the European organic chemical synthesis revolution from the mid-19th century (e.g. diacetylmorphine 1874) to the early 20th century (e.g. oxycodone in 1916) in the now seemingly vain search for opioid analgesics without addictive effects, we now have over 30 opioids on the market worldwide. These can be broadly categorised into three chemical groups: 4,5-epoxymorphinans (e.g. morphine), 4-phenylpiperidines (e.g. fentanyl), and 3,3-diphenylpropylamines (e.g. methadone). Since the early 1960s, there have been very few new opioids marketed, but there have been many new and novel formulations, mainly because the ‘holy grail’ of pain relief without harmful effects (e.g. dependence, respiratory depression or the more encompassing opioid-induced ventilatory impairment) had not been achieved. This was based on the false belief that all opioids are essentially the same, so choosing the cheapest opioid for treatment was a rational approach. However, as with many old drugs, modern drug development bypassed these opioids, and we are now gaining a better understanding of key differences in their pharmacology. For example, QT prolongation and drug metabolism polymorphism were never part of the requirement for the regulatory approval of opioids, requiring researchers to make these discoveries and clinical recommendations in lieu of the pharmaceutical industry. The past 20 years has seen a revolution in our understanding of opioid drug action mainly driven by the great advances in molecular biology and pharmacology.
In this overview, we will review how these advances have impacted opioid pharmacology, from atypical opioids currently on the market to those in the pipeline, to the contentious biased mu agonist concept, mixed agonists, the ‘rediscovery’ of the role of delta and kappa receptors, to heterodimers and to genetics (including splice variants) and interactions with the immune system. Often, opioids have been the model substrates for contemporary basic and experimental investigations, mainly because of the potential translation to achieve the ‘holy grail’. Where possible, the clinical context and relevance have been briefly stated.
Atypical opioids
Typical opioids such as morphine, oxycodone, and fentanyl almost exclusively act on the mu opioid peptide (MOP) receptor. Although several old opioids are in reality ‘atypical opioids’, there has been renewed research into developing new opioids with an ‘atypical’ badge, mainly due to recognition that pain pathways comprise multiple biochemical elements and that addressing more than one element (e.g. MOP receptor) should improve efficacy but not necessarily safety. Some have been registered and others are in development. Here we review atypical opioids from a pharmacological and clinical perspective.
Tramadol
Tramadol was first introduced into the German market in the mid-1970s, and is the first truly developed atypical opioid. It has weak MOP receptor activity, requiring polymorphic CYP2D6 metabolism to the 400-fold more opioid active O-desmethyltramadol metabolite. In contrast, tramadol itself has inhibitory activity towards the serotonin transporter SLC6A4 (SERT) and norepinephrine (noradrenaline) transporter SLC6A2 (NET), which particularly affects the descending inhibitory pain pathway.1,2
Methadone
Methadone was developed in Germany during the Second World War and the MOP receptor has a high nanomolar (nM) affinity to methadone with the R enantiomer having a higher affinity than the S enantiomer. The opposite occurs for its interactions (S > R) with subunits of the ionotropic glutamate N-methyl-
Buprenorphine
Buprenorphine was developed in the 1960s and is a very potent opioid with very high binding affinity for all three opioid receptors, mu (MOP), delta opioid peptide (DOP) and kappa opioid peptide (KOP). Its major metabolite, norbuprenorphine, has a similar high affinity to the MOP receptor but eight-fold less for delta and kappa receptors. Buprenorphine is often considered a partial agonist, mainly based on animal analgesic studies (an antagonist of delta and kappa receptors) but in humans, it acts more as a full agonist for analgesic effects and a partial agonist for respiratory effects, requiring larger and more prolonged naloxone doses to reverse respiratory depression.5,6
Tapentadol
Tapentadol has activity at the MOP receptor but its affinity of 160 nM is about 20-fold greater than for morphine, and it has weak inhibitory action and binding to NET. 7 These ‘relatively weak’ actions at the two targets are considered to produce a synergistic interaction for pain relief. Raffa and colleagues 8 have calculated that the ‘µ-load’ was less than 40% compared to 100% for ‘pure’ opioids. Clinically this appears to translate into less severe gastrointestinal effects from immediate-release acute pain therapy, 9 and lower odds of causing typical opioid adverse effects including respiratory depression and dependence.10,11 Given its place in pain treatment is still being investigated, it remains too early to assign its advantages over ‘pure’ opioids such as morphine and oxycodone.
Cebranopadol
Cebranopadol is a mixed MOP receptor and nociceptin/orphanin FQ (F=phenylalanine; Q=glutamine) peptide (NOP) receptor (see below) full agonist having an affinity for NOP and MOP receptors of about 1 nM. 12 It is undergoing clinical trials for chronic pain, and for use in cancer pain over 26 weeks results indicate that a dose of up to 1 mg/day has some efficacy and adverse effects that are opioid related (e.g. gastrointestinal). 13
In summary, we reached the full opioid agonist class of drugs over 50 years ago, but we now have some opioids which are atypical in that they have affinity for other targets at therapeutic concentrations, such as transporters (SERT, NET), and non-mu receptors (NOP, NMDA), all of which are associated with pain pathways and hence these could result in clinical synergy with the mu agonist component of the drug. Nevertheless, they will all show the typical major mu opioid adverse effects (e.g. respiratory depression, tolerance and dependence) depending on dose and patient characteristics. Hence careful selection of patient pain phenotype (nociceptive–neuropathic–nociplastic) and dose optimisation is likely to be warranted.
Biased mu agonism
MOP receptor agonists such as morphine are highly effective for the treatment of severe pain and are clinical mainstays for pain management. 14 However, deaths due to overdose-induced respiratory depression have occurred on numerous occasions,15–18 and their euphoric effects present a liability for the development of addiction. As mentioned, common clinical opioids exert analgesic effects by binding to endogenous MOP receptors in neuronal membranes to trigger signalling cascades. These cascades begin with G-protein activation and modulation of membrane ion channel behaviour along nociceptive pathways and central pain-processing centres to provide analgesia. However, prolonged activation of these receptors leads to recruitment of β-arrestin-2 (βarr2) which inhibits continued G-protein signalling, subsequently activating protein kinase cascades that can result in other opioid receptor—facilitated effects. Knockout animal studies have shown that recruitment of βarr2 underlies side-effects associated with mu opioid agonism including respiratory depression, constipation, nausea and addiction (Figure 1).19–21 The coupling of MOP receptors to multiple signalling pathways makes them ideal targets for biased ligands that may favour G-protein interaction over recruitment of βarr2. 22 In their seminal review, Gillis et al. 23 define a biased agonist as one that ‘… activates one signalling pathway downstream of a receptor more efficiently than it activates another, discrete signalling pathway of the same receptor … in comparison to reference ligands’. This has driven the research and development of biased MOP receptor ligands in an attempt to provide better, more sustainable analgesia devoid of serious adverse effects, and several drug candidates have already been investigated.24–26
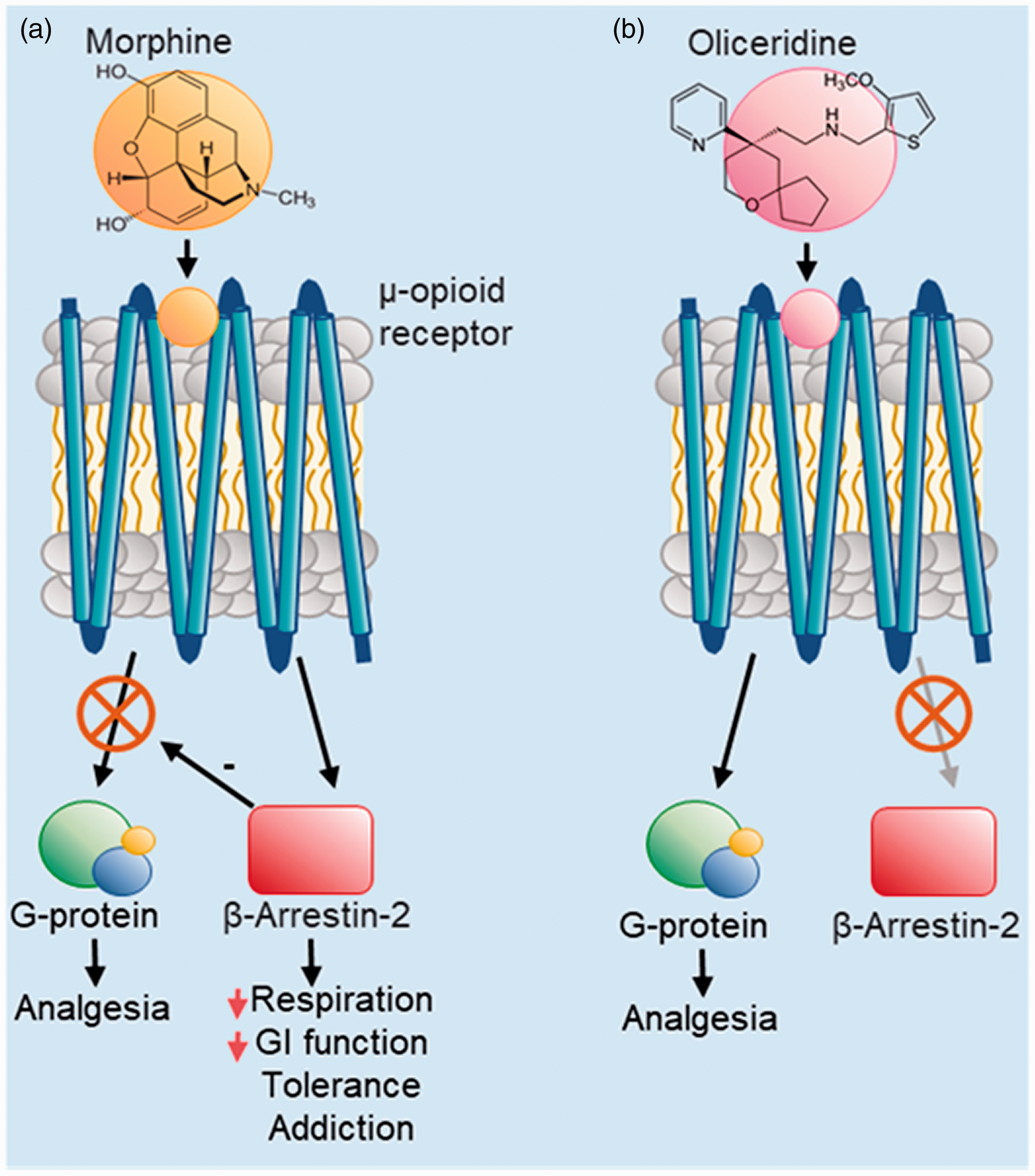
Bias of mu opioid peptide (MOP) signalling pathways correlates with the analgesic and side-effects of opioids. (a) MOP receptor agonism by unbiased compounds such as morphine trigger signalling through both G-protein and β-arrestin-2 pathways. (b) Biased compounds such as oliceridine are intended to trigger the G-protein—mediated signalling to produce analgesia without side-effects caused by the β-arrestin-2 pathway.
Oliceridine
Oliceridine (also known as TRV130) is a central nervous system (CNS)-active G-protein—biased ligand that binds to the MOP receptor with a low nM affinity and provides similar maximal analgesic effects to morphine with higher potency in animal studies.27,28 When assayed in vitro, oliceridine demonstrated G-protein signalling bias, reduced recruitment of βarr2, and reduced MOP receptor internalisation compared to morphine.24,27 Similarly, in vivo it provided dose-dependent antinociception with similar maximum effects to morphine with approximately ten-fold greater potency and an improved side-effect profile.24,29 These findings have since been replicated, confirming the G-protein bias and antinociceptive action of oliceridine, and its favourable tolerance development compared to morphine 30 and fentanyl. 31 In contrast, others have shown gastrointestinal inhibition and abuse liability can still arise following repeated oliceridine administration similar to non-biased opioids such as fentanyl and oxycodone.32–34 Many phase II and phase III clinical trials have shown oliceridine to provide equal or better analgesic effects than morphine for moderate to severe acute pain, with reduced adverse effects such as nausea, tolerance, gastrointestinal inhibition and respiratory depression.25,26,35–38 A phase III multicentre study of 718 patients reported oliceridine to provide a rapid reduction in the numerical pain score within 30 minutes of administration. 39 Common adverse effects reported included nausea (31%), constipation (11%) and vomiting (10%); however, 97% of patients reported none to mild withdrawal symptoms, and respiratory depression was found to be significantly lower when compared to morphine in a retrospective observational study. 40 Despite these findings, the US Food and Drug Administration approval for oliceridine was initially rejected in 2018 following concerns with dosing strategies aimed to treat moderate to severe pain, and side-effects associated with high doses. Following a 2020 resubmission, oliceridine (Olinvyk[insert trademark sign], Trevena, Inc., Chesterbrook, PA, USA) was approved for short-term intravenous usage in hospitals and other controlled settings when alternative treatment options are inadequate, although it is distributed with a boxed warning concerning abuse, misuse and potentially fatal respiratory depression. Although phase II and phase III trials have established oliceridine to provide potent analgesic effects, more definitive data are needed to demonstrate its improved respiratory and gastrointestinal profile compared to morphine. 41 Further investigations are also recommended to understand better its safety in high-risk populations, and in patients with comorbidities such as chronic obstructive pulmonary disease, renal and hepatic impairment.
Other compounds in development
PZM21 is another biased ligand discovered by using information on over three million commercially available compounds which dock to the MOP receptor binding pocket. 42 In receptor binding studies it showed high affinity for MOP receptors and weak antagonistic KOP receptor activity. Functional studies revealed it to be a potent agonist in many G-protein—mediated signalling assays, while effectively having no impact on βarr2 recruitment. 43 Preclinical studies involving PZM21 have reported analgesia devoid of respiratory depression and G-protein activation bias similar to oliceridine. 43 However, there is also evidence to suggest it has low efficacy in G-protein activation, produces tolerance and depresses ventilation in rodents similar to classic opioids such as morphine.44,45 Similar results were found in non-human primates with dose-dependent antinociceptive effects of PZM21 being tenfold less potent than oxycodone while exerting similar reinforcing effects. 46
SR17018 was one of a series of biased MOP receptor agonists tested in vivo for antinociceptive effectiveness versus ventilatory suppression. It displayed the highest G-protein signalling bias and ED50 for respiratory depression among all compounds, and demonstrated a positive correlation between G-protein signalling bias, decreased ventilatory suppression and therapeutic efficacy. 21 Compared to drugs such as fentanyl and SR11501 that exhibit an arrestin signalling bias, SR17018 showed a therapeutic window 25-fold higher.21,47 Even at a high dose, SR17018 did not activate βarr2 signalling in vitro or block its recruitment by other MOP receptor agonists, suggesting that its administration may stabilise a configuration of the MOP receptor that has no affinity for βarr2.21,48
SHR9352, a compound made through structural changes to oliceridine, has demonstrated high in vivo efficacy with high affinity for MOP receptor compared with delta and kappa receptors. Similar to oliceridine, it has a strong G-protein activation bias and limited βarr2 recruitment. 49
Mitragynine pseudoindoxyl is naturally derived from the Mitragyna speciosa plant with antinociceptive properties that have been investigated both in vitro and in vivo.50,51 It is a partial MOP receptor agonist, and a competitive antagonist at kappa and delta opioid receptors that has also demonstrated G-protein bias.52,53 In rodent studies, it provided potent antinociception devoid of reward, tolerance, respiratory depression and gastrointestinal inhibition, 53 demonstrating that biased MOP receptor agonism combined with kappa and delta opioid receptor antagonism may underlie opioid-induced analgesia without side-effects. 53 In humans, however, mitragynine has emerged as a drug of abuse. The risk posed by the drug from case studies involving mitragynine-related fatalities remains uncertain as simultaneous drug use prior to death is often reported.54,55
In summary, highly selective MOP receptor agonism is effective in producing clinical analgesia, although it is also responsible for the side-effect profile associated with clinical opioids. G-protein bias at the MOP receptor has been widely asserted to characterise a novel and improved class of opioid analgesic based on the hypothesis that βarr2 signalling underlies the adverse effects of MOP receptor agonism.19–22 However, this idea has recently been challenged23,32 by studies in βarr2 knockout mice, 56 and mice expressing G-protein—biased MOP receptor 57 demonstrating failure of improvement in the therapeutic index of morphine. In addition, re-evaluation of lead G-protein—biased ligands showed they exhibited low intrinsic efficacy compared to morphine, 58 suggesting low-efficacy partial agonism may be responsible for improved therapeutic indices of these compounds rather than G-protein bias.23,58 The use of structure-based drug screening and computational docking has been very effective in the identification and creation of biased MOP receptor ligands, and new drugs currently in development have demonstrated higher degrees of G-protein bias compared to oliceridine. However, more thorough research is needed to confirm the contribution of G-protein bias to the potentially improved therapeutic index provided by these compounds, along with rigorous animal and human in vivo testing to understand their potential for abuse. Future studies that link in vitro signalling pathway events and MOP receptor conformations during ligand binding to in vivo behaviour are also needed to understand better the differential effects of these compounds compared to unbiased opioids. The use of varied animal models during these assays must also be considered as the analgesic and side-effect profiles arising from MOP receptor agonism differ greatly between rodent models and primates.46,59–61
Finally, from a thoughtful clinical perspective, Ballantyne and Chavkin in their topical review, 60 in which they focused on the adverse effects of respiratory depression, abuse potential and analgesic tolerance, concluded that it was ‘unclear’ whether ‘biased ligands will be the opioids of the future’. Indeed, unless there are multiple breakthroughs in opioid receptor pharmacology, it is very unlikely that a ‘pure’ MOP receptor biased ligand will suffice, and mixed/atypical ligands targeting other receptors (opioid and non-opioid) may be more fruitful as is the case of many other drug treatments of chronic conditions (e.g. cardiovascular, infectious disease, psychiatric).
Other opioid receptors
Although the MOP receptor is the primary receptor to be targeted for the relief of moderate to severe acute and chronic pain, some of the other known opioid receptors have analgesic and conversely adverse effects which need to be considered in the individual patient as all opioids target these other receptors to varying degrees.
The NOP receptor is considered the fourth opioid receptor whose function remains to be extensively investigated. It is found in the same neuronal pathways as opioid receptors and inhibits neuronal transmission. In animals, activation causes antinociceptive effects spinally, supraspinally and systemically, 62 and when combined with opioids has a synergistic effect on antinociception. NOP agonists have neither respiratory depressant nor reinforcing effects, the latter highlighting the potential for non-addictive behavioural effects. Full co-agonists of both MOP and NOP receptors, which have been developed, are likely to have these effects. Attempts are being made to generate not full but partial agonists such as AT-121, which in animal studies does not induce these unwanted ‘mu opioid’ effects. In their study, Ding et al. suggested that ‘bifunctional NOP/MOP agonists with the appropriate balance of NOP and MOP agonist activity may provide a dual therapeutic action for safe and effective pain relief and treating prescription opioid abuse’. 62 Other similar partial agonists such as BU08028 and BU10038 are in development. 63
Recently DOP has attracted interest in the field of acute migraine. DOP is structurally closely related to the MOP receptor but its cellular distribution remains to be firmly delineated. 64 DOP activation appears to cause less respiratory depression, gastrointestinal motility disturbance and sedation than that for mu receptor activation, and its euphoric effects and physical dependence propensity are also minor. 65 TRV250 is a small-molecule DOP agonist of preferential selectivity for G-protein signalling and targeted for acute migraine, and a first-in-man study has been published. 66 It is also being trialled for other pain conditions such as acute and chronic pain (knee osteoarthritis), postherpetic neuralgia and depression, 67 but evidence of efficacy in these areas seems to have resulted in abandonment of further investigations.
KOP activation produces some degree of peripheral analgesia but produces dysphoria and hallucinations 65 and diuresis. 68 Hence there is minimal if no current interest in development for therapeutics.
In summary, from a clinical perspective it is unlikely that we will see any new opioids that solely target these receptors.
Other pain targets
The expression, signalling and desensitisation of opioid receptors is affected by their interactions with other G-protein—coupled receptors (GPCRs) such as chemokine receptors, which play a major role in many processes including inflammation through altering immune cell migration.69,70 Opioid peptides activate ACKR3 (a member of the atypical chemokine receptor family and formerly known as CXCR7), which appears to be a negative regulator of opioid peptide (mainly dynorphin) function and is proposed as an opioid scavenger receptor. 69 Understanding of its role in opioid action is still very much in its infancy but does highlight the likelihood that future drug development of pain therapeutics will include an array of drugs targeting receptors often not considered important, particularly in the field of chemokine pharmacology.
FF3 is an opioid analogue of fentanyl designed to reduce pain by activating, in a selective manner, peripheral MOP receptors in inflamed, that is acidic, tissues. 71 In rat models of various pain phenotypes (e.g. postoperative, neuropathic) given at low doses, it elicited the desired effects, but at higher doses it produced the typical opioid effects of sedation, respiratory depression, reward and constipation. Pharmacokinetic studies are clearly needed, especially if transdermal formulations are being considered for local analgesia.
ZH853, an endomorphin analogue, does not elicit pro-inflammatory effects (as opposed to most opioids—see below), has a long duration of action in aimals, and has been shown to have reduced effects on abuse liability, respiratory sequelae, motor impairment, and glial activation.72 This was based on the findings of Grace et al., 73 who showed that in animal models of neuropathic pain, morphine prolongs pro-inflammatory signalling in the spinal cord dorsal horn. Feehan and Zadina, using an animal model of chronic pain, showed that ZH853 reduced both the time spent in chronic pain and pain severity compared to morphine, and proposed clinical development ‘for inflammatory and postoperative pain’. 74 This highly interesting area of glial activation, pro-inflammation and opioids has opened up new vistas as to how we view opioids, especially from a chronic clinical pain context and perspective.
Splice variants and multiple isoforms
This is an exciting new area of opioid drug discovery and development that may provide the clinician with a greater understanding of opioid actions (good and bad) in the individual patient.
During transcription, genes that encode GPCRs can be processed differently, resulting in multiple versions of the final messenger RNA, called splice variants. 75 This can result in specific domains of the receptor being missing or abnormal domains being added, resulting in a range of isoforms, each potentially activating a different downstream signalling pathway. Added to this complexity is that the range of isoforms can be tissue specific. Marti-Solano et al., 76 through a series of elegant studies, showed that almost half of the human GPCRs that are targets of US Food and Drug Administration—approved drugs have more than one isoform, and in many instances each of the isoforms for a given receptor has a different tissue distribution. MOP receptor isoforms from splice variants have been known for over 20 years. 77 Marti-Solano et al. showed that OPRM (opioid receptor mu; the mRNA of the MOP receptor) had three unique combinations of isoforms across four different human tissues. These findings also raised the potential of MOP receptor isoform expression in different tissues and sites (e.g. neurons in different sites within the CNS, glia) to change over time (e.g. acute to chronic pain) and therefore to display different phenotypes (nociceptive versus neuropathic versus nociplastic versus combination). Thus, opioid drugs may need to be both receptor subtype (e.g. MOP, DOP), and isoform selective. 75 Translating this clinically for the individual pain patient would represent the pinnacle of precision medicine for pain relief.
Heterodimers
GPCRs such as the MOP receptor can exist as a homodimer (e.g. two MOP receptors linked) but also as a heterodimer (two different receptors linked such as MOP/NOP or MOP/cannabinoid receptor type 1 (CB1)). They have been constructed in cell lines and in vitro functional activity has been obtained. For example, the DOP component of MOP/DOP appears to exert an antagonistic effect on MOP receptor function and may have distinct intracellular signalling and trafficking outcomes and co-expression in specific neuronal populations in the spinal cord, but not in other sites. 78 Chronic morphine use appears to upregulate the expression of the MOP/DOP heterodimer. 79 Compound CYM5010 that targets the heterodimer was analgesic in various animal models including for neuropathic pain, and also showed less tolerance, but its potential clinical development is not apparent. There are also MOP/KOP heterodimers and heterodimers with other receptors such as with galanin receptor 1 and CB1, 80 but whether specific ligands and clinical translation can be developed appears to remain in the far distant future.
Opioimmunomodulation
In addition to opioid activity at neuronal opioid receptors, there is now extensive evidence that opioids interact with our immune system. Early indications of an opioid-immune interaction came from observations of apparent opioid immunosuppressive effects on the peripheral immune system. 81 Conversely, immunocompetent cells of the brain and spinal cord, the glia (astrocytes and microglia), are activated following prolonged exposure to opioids. Extensive preclinical evidence now supports that this opioid-induced glial activation and production of pro-inflammatory mediators contributes to opioid-induced tolerance, hyperalgesia and allodynia. This suggests an opportunity to separate the negative effects of opioids from their analgesic effects, either via novel opioid pharmacology (e.g. opioid agonists with reduced innate immune activation potential) or adjuvant therapy (e.g. modulation of relevant immunology).
Opioid interactions with immune cell receptors
MOP, DOP, KOP and NOP receptor mRNA have been detected in human peripheral immune cells, although not consistently, and at low levels relative to neuronal tissue, with less support for expression on glia. 82 Evidence of functional effects of opioids at classic opioid receptors in primary immune cells is also equivocal, while opioid receptor triple (mu, delta, kappa) knockout mice still develop hyperalgesia with sustained morphine, oxymorphone or fentanyl exposure, indicating an opioid receptor—independent mechanism.83,84 This is supported by in vitro studies demonstrating that structurally diverse opioids (including the clinically relevant agonists morphine, fentanyl, remifentanil, methadone, oxycodone, buprenorphine, meperidine and antagonists naloxone and naltrexone) interact with Toll-like receptor 4 (TLR4) (an innate immune pattern recognition receptor with a major role in central immune mechanisms of pathological pain) 85 and/or its accessory protein myeloid differentiation protein 2 (MD2).86–89
MOP agonists and antagonists tend to be TLR4/MD2 agonists and antagonists, respectively; however, the potencies are not necessarily correlated. For example, morphine-3-glucuronide displays significant TLR4 activity, but no opioid receptor activity, while the opioid active morphine-6-glucuronide is devoid of TLR4 activity (Figure 2). 87
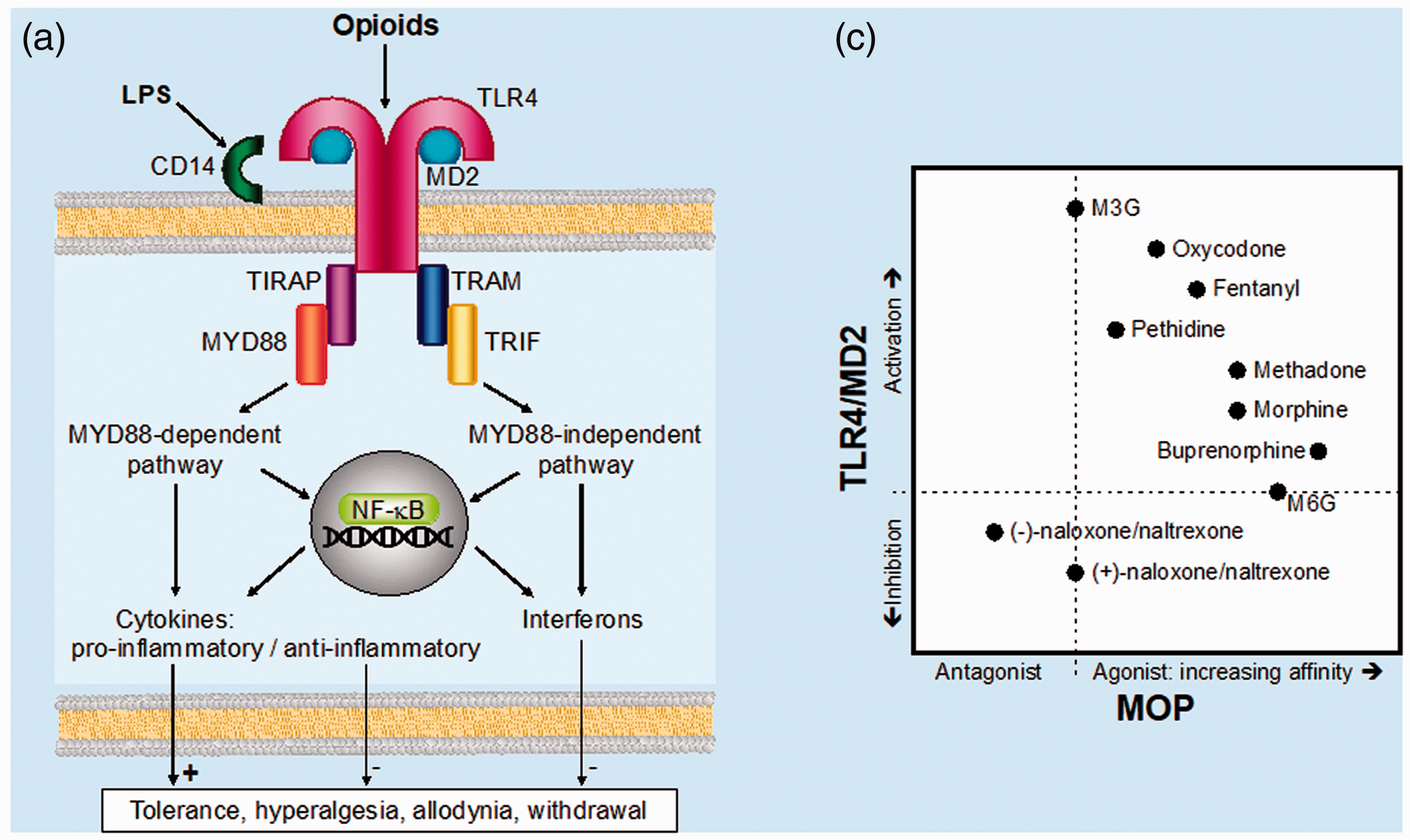
(a) Opioid interactions with glial Toll-like receptor 4 (TLR4)/myeloid differentiation protein 2 (MD2), consequent intracellular signalling cascades and cytokine release proposed to contribute to opioid-induced analgesic tolerance, hyperalgesia, allodynia and withdrawal. (b) Mu-opioid peptide (MOP) receptor agonist (rank-order in vitro affinity(unpublished)) and antagonist versus rank-order in vitro TLR4/MD2 activation and inhibition of different opioids. 86 LPS: lipopolysaccharide (endotoxin); M3G and M6G: morphine-3-glucuronide and morphine-6-glucuronide, respectively; TIRAP: Toll-interleukin-1 receptor (TIR) domain-containing adaptor protein; TRIF: TIR domain-containing adaptor protein-inducing interferon-beta; TRAM: TRIF-related adaptor molecule; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells.
Importantly, opioid agonists and antagonists act non-stereoselectively at TLR4/MD2 (compared to (–)-isomer selectivity of classical opioid receptors), providing an opportunity for the use of (+)-isomers of opioid antagonists (e.g. (+)-naloxone, (+)-naltrexone) to block the negative effects of opioimmunomodulation while maintaining MOP receptor activity and analgesia.87,88,90
In addition to high expression on glia, particularly microglia, TLR4 is found in rat isolated dorsal root ganglion neurons 91 and primary rat CNS endothelial cells. 92 Whether opioids interact directly with other non-opioid innate immune cell receptors (e.g. other pattern recognition receptors such as TLR2), and the degree of cross-talk between MOP and TLR4 signalling, is an area of open investigation.93,94
Preclinical in vivo opioimmunomodulation
In rodents, morphine exposure (three to nine days) results in significant activation of glial cells and increases in spinal cord and brain expression of key innate immune signalling receptors and pro-inflammatory cytokines.93,95–106 While a major component of this immune activation is likely to be via direct actions of opioids on immune cells, there is preclinical evidence that neuronally derived peptides and chemokines can also contribute to indirect glial activation and tolerance following morphine administration.107–109 Regardless of whether opioid activation of innate immune cells is direct or indirect, there is now extensive preclinical evidence that inhibition of this activation or subsequent pro-inflammatory signalling can mitigate multiple negative effects of repeat opioid exposure. For example, pre- or co-administration of TLR4/MD2 antagonists, glial cell inhibitors or pro-inflammatory cytokine signalling inhibitors, as well as anti-inflammatory signalling enhancement, attenuates the development of analgesic tolerance, hyperalgesia, allodynia and withdrawal behaviours following repeated morphine exposure in rodent nociceptive pain86,87,95,98–106,110,111and nerve injury88,101,103,112,113 models.
TLR4/MD2 antagonists, TLR4 genetic knockout, glial cell inhibitors and inhibitors of spinal cytokine signalling, also potentiate acute morphine analgesia in rodent nociceptive pain models,86,87,104–106,114,115 supporting a role for opioimmunomodulation in acute opioid analgesia.
Preclinical research in this area has focused almost entirely on morphine; however, as expected from in vitro data, 87 single studies of methadone, 105 fentanyl, 116 and oxycodone 116 demonstrate similar potential to morphine for the development of analgesic tolerance, hyperalgesia and allodynia, or exacerbating nociceptive hypersensitivity in rat models of nerve injury. Furthermore, glial, TLR4 and cytokine inhibition potentiated acute methadone, 105 oxycodone 104 and remifentanil 89 analgesia. Therefore, opioid-immune interactions are not just restricted to morphine and the 4,5-epoxymorphinans.
Clinical evidence for potential therapeutic improvement through modified opioimmunomodulation
Directly demonstrating the importance of opioimmunomodulation to clinical pain management is in its early stages. Innate immunoreactivity of peripheral blood immune cells is significantly modified in chronic pain patients on opioids, compared to chronic pain patients not on opioids and pain-free controls. 117 Clinical genetics studies have also identified polymorphisms in innate immune signalling pathway genes that contribute to interpatient variability in pain and opioid response in postoperative and cancer pain patients.118,119
Disparities in agonist/antagonist structure–activity relationships and potencies for MOP receptors versus TLR4/MD2 have raised the possibility of developing novel opioid analgesics that maximise MOP receptor, and minimise TLR4/MD2, activity. 87 However, this has yet to be realised. There is also the potential to mitigate the immunopharmacology of current opioids via adjuvants that block opioid central immune activation. Several TLR4 antagonists and glial modulators shown to reduce opioid-induced tolerance, hyperalgesia and allodynia preclinically are clinically available for other indications and as such have the potential to be repurposed. Some have been trialled, or are undergoing clinical trials, as potential novel analgesics in their own right, or to prevent persistent postoperative pain. However, aside from low-dose naltrexone, which has demonstrated some success in fibromyalgia, they (e.g. minocycline, propentofylline) have so far shown only limited efficacy. 120 In terms of improving the clinical efficacy of opioids, ultralow-dose naltrexone reduces postoperative opioid requirements and opioid-related side-effects. 121 Pre-surgery pentoxifylline also decreases postoperative opioid requirements,122,123 while ibudilast has shown some potential for reducing opioid withdrawal symptoms and enhancing the analgesic effects of oxycodone in opioid-dependent patients.124,125
Conclusion
The search for new opioids with reduced adverse effects, especially opioid-induced ventilatory impairment and addiction, has continued unabated over the past decade following the hiatus in the late 20th century. This has come about by a greater understanding of the molecular pharmacology of these drugs driven by the genomic revolution. It is now apparent that not all opioids are the same. The future is likely to see more atypical opioids, biased agonism being better studied as we gain a greater understanding of intracellular events, sophisticated and novel heterodimer drugs, and exploitation of the contribution of opioimmunopharmacology to current opioid actions. Other effects such as opioid-induced hyperalgesia 126 and endocrine dysfunction 127 have not been investigated clinically with many of the newer analgesic drugs. This may be due to these effects occurring after longer periods of dosing than in the current trials, and also that they may not be considered as important as the respiratory and addictive effects. Hopefully, the fine tuning in analgesic drug action and development described in this overview will allow for better precision medicine in acute and chronic pain management with less patient harm.
Footnotes
Author Contribution(s)
Declaration of conflicting interests
The author(s) have no conflicts of interest to declare.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.