
Abstract
This narrative review aims to describe the role of peripheral and central immune responses to tissue and nerve damage in animal models, and to discuss the use of immunomodulatory agents in clinical practice and their perioperative implications. Animal models of pain have demonstrated that nerve injury activates immune signalling pathways that drive aberrant sensory processes, resulting in neuropathic and chronic pain. This response involves the innate immune system. T lymphocytes are also recruited. Glial cells surrounding the damaged nerves release cytokines and proinflammatory mediators that activate resident immune cells and recruit circulatory immune cells. Toll-like receptors on the glial cells play a crucial role in the pathogenesis of chronic pain. Animal models indicate an immune mechanism of neuropathic pain. Analgesic drugs and anaesthetic agents have varied effects on the neuroimmune interface. Evidence of a neuroimmune interaction is mainly from animal studies. Human studies are required to evaluate the clinical implications of this neuroimmune interaction.
Introduction
Many rodent models of tissue and nerve injury have demonstrated the link between the nervous system and the immune system. 1 Nerve injury disrupts the primary sensory neurons and the central projection pathways and activates immune responses throughout the pain pathways as well. Damaged nerves and surrounding glial cells release mediators which activate resident immune cells and recruit more immune cells from the systemic circulation. The immune cells then release cytokines, chemokines and pro-inflammatory mediators which alter the transmission of pain signals at the nerve trunk, cell bodies of the dorsal root ganglion and synaptic terminals in the dorsal horn of the spinal cord (Table 1). However, these animal models have limitations. It is not easy to assess what is actually perceived by the animal. Changes in cutaneous sensory thresholds in response to nerve injury rather than integrated pain behavioural responses are measured. These animal models of pain (Table 2) develop spontaneous pain behavioural changes, allodynia and hyperalgesia measured by altered thresholds to mechanical (using a von Frey hair) and sensory (e.g. cold) stimuli.
Sequence of neuroimmune activation in neuropathic pain.
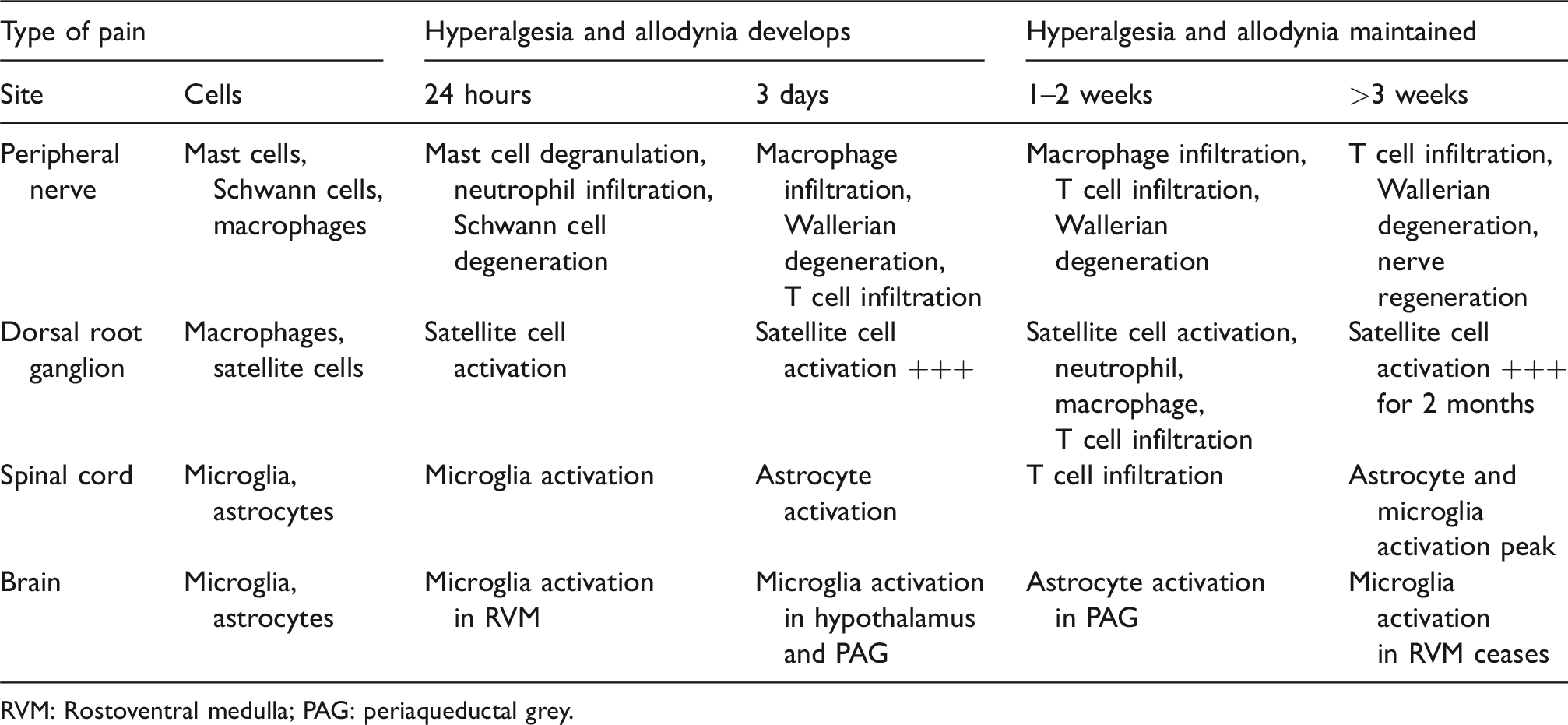
RVM: Rostoventral medulla; PAG: periaqueductal grey.
Some animal models of neuropathic pain.
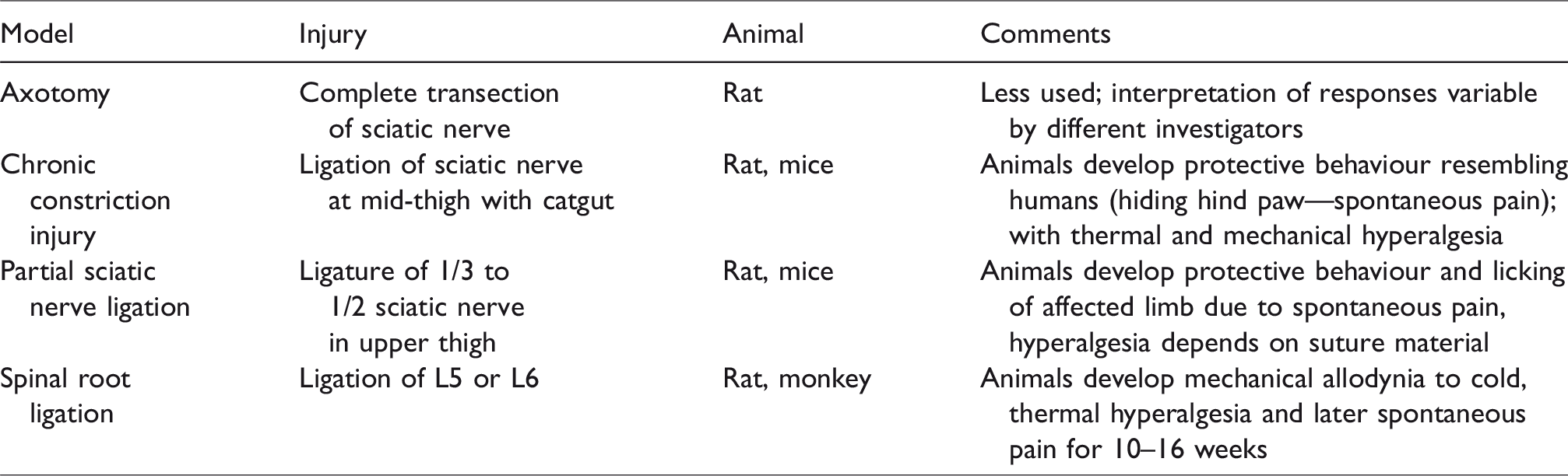
The aims of this narrative review are to describe the immune cellular responses to nerve injury that contribute to pain, and to explore the use of immunomodulatory therapeutic agents in clinical practice.
Search strategy and selection criteria
A literature search was conducted using MEDLINE (PubMed), EMBASE and CINAHL databases using the following key words and MESH terms: neuropathic pain, immune function, cytokines, lymphocyte function, anaesthesia, analgesia, opioids, local anaesthesia, non-steroidal anti-inflammatory drugs, tricyclic antidepressants and a combination of those. The search was limited to English language articles published between January 1990 and March 2018. We manually searched the reference lists of the articles to identify further relevant articles. The initial data retrieval was performed independently by both authors, and the resulting data-set was agreed by discussion between the two authors. A total of 806 articles on the neuroimmune mechanisms of pain were retrieved. From these, 106 original and review articles were selected. Human studies were examined, and the levels of evidence were graded.
The immune response to injury
Macrophages, leukocytes, mast cells, glial cells and T lymphocytes are immune cells involved in the peripheral and central pain pathways. In response to tissue damage and nerve injury, these cells are activated and release inflammatory mediators and cytokines in the skin, peripheral nerves, dorsal root ganglia and spinal cord 2 (Figure 1).
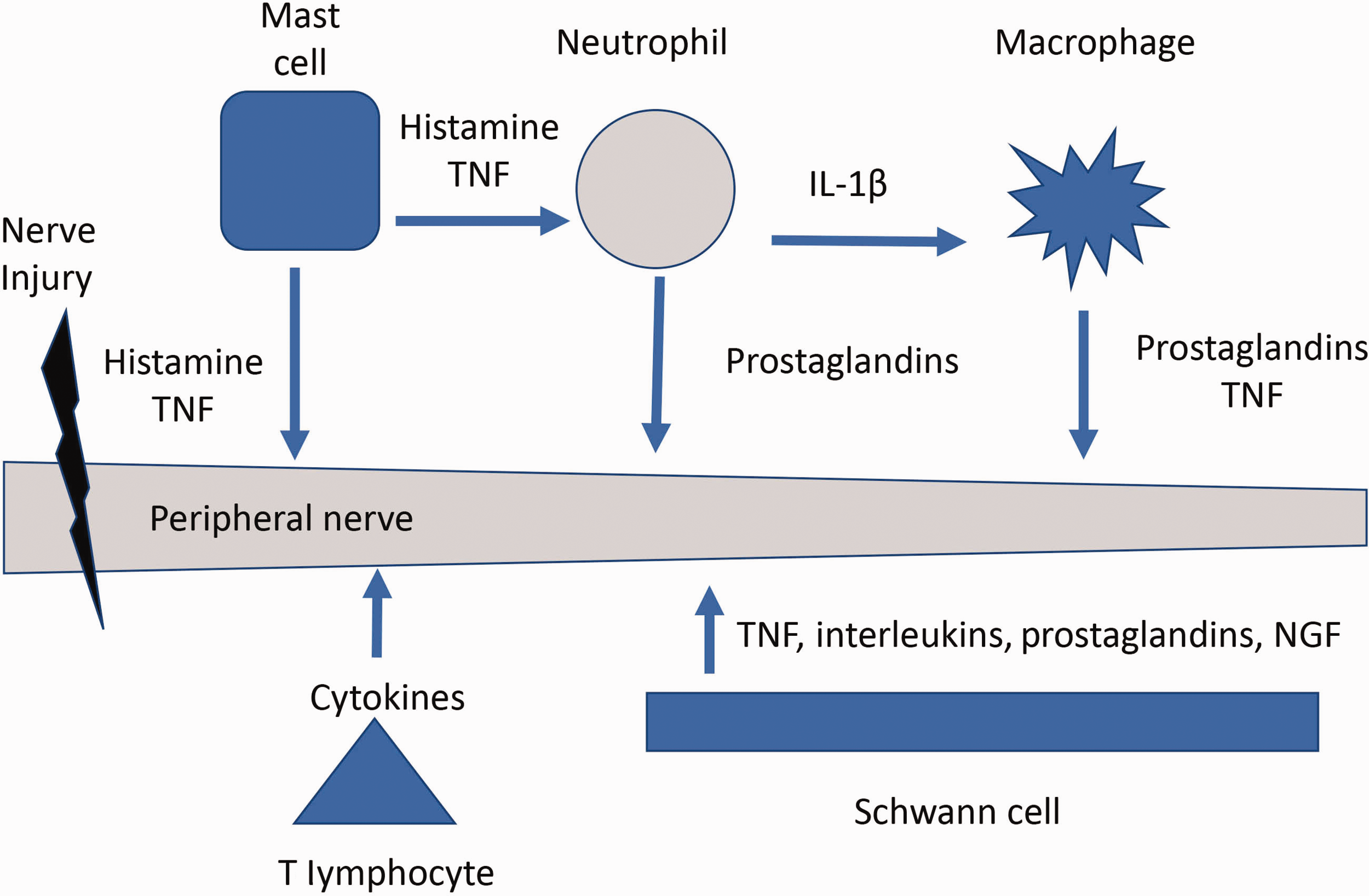
Activity of neuroimmune cells in peripheral nerve injury. TNF: tumour necrosis factor; IL-1β: interleukin-1β; NGF: nerve growth factor.
Cytokines (tumour necrosis factor (TNF), interleukins (IL-1β, IL-6)), adenosine triphosphate (ATP), brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) are released by activated immune and glial cells (Schwann cells, microglia, satellite cells and astrocytes). These immunological responses are involved in the pathogenesis of neuropathic and chronic pain 3 (Table 3).
Peripheral somatosensory nerves: activation of immune cells.
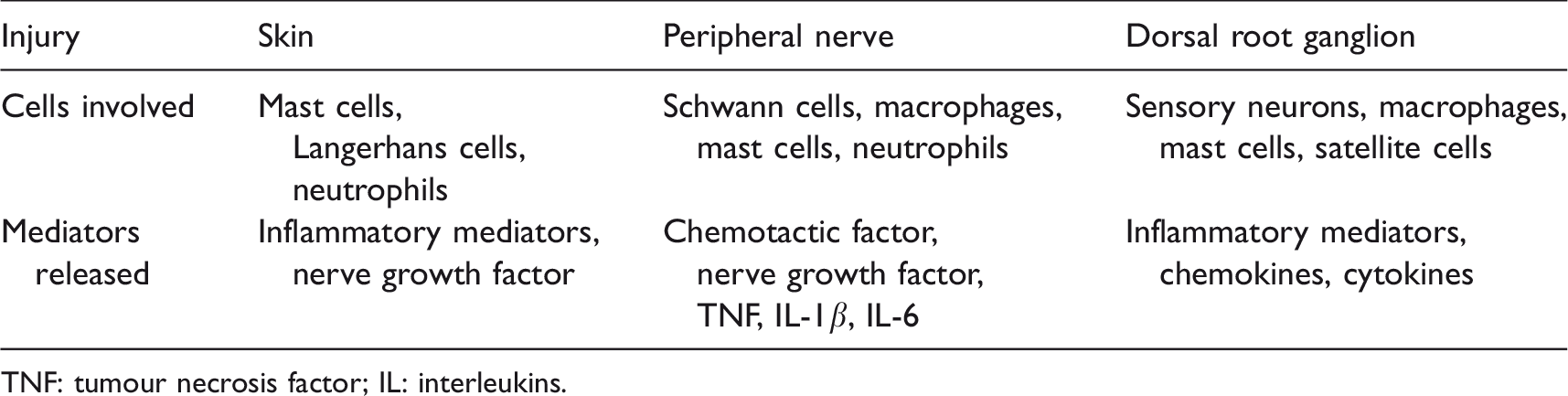
TNF: tumour necrosis factor; IL: interleukins.
Mast cells
Mast cells are important initiators and effectors of innate immunity. When the primary afferent nociceptors in the skin are damaged, mast cells, keratinocytes and Langerhans cells that lie in close proximity to sensory afferent axons are activated. The activated mast cells around the peripheral nerves degranulate and release mediators such as histamine, TNF, serotonin, tryptase, transforming growth factor β (TGF-β) and leukotrienes at the site of nerve injury2,3 (Table 4). Mast cell degranulation can be demonstrated in animal models of pain (trigemino-cervical, lumbosacral and chronic visceral pain).4–6 Histamine released by mast cells sensitises peripheral nociceptors. Spontaneous pain is increased by histamine applied topically on the skin of post-herpetic neuralgia patients. 4 Histamine receptors on the nerves are up-regulated following nerve injury and mediate neuropathic pain. 5 In addition, mast cells cross the blood–brain barrier and facilitate the spread of pro-inflammatory changes into the central nervous system (CNS). 6 Histamine antagonists have been found to reduce mechanical and thermal hypersensitivity after partial sciatic nerve ligation (PSL) in a rat model. 5 It has been reported that diphenhydramine and hydroxyzine have mild analgesic properties in trigeminal neuralgia, thalamic pain syndrome, refractory cancer pain and dysmenorrhoea5,6 (Level 3 evidence).
Neuroimmune mediators of mast cells.
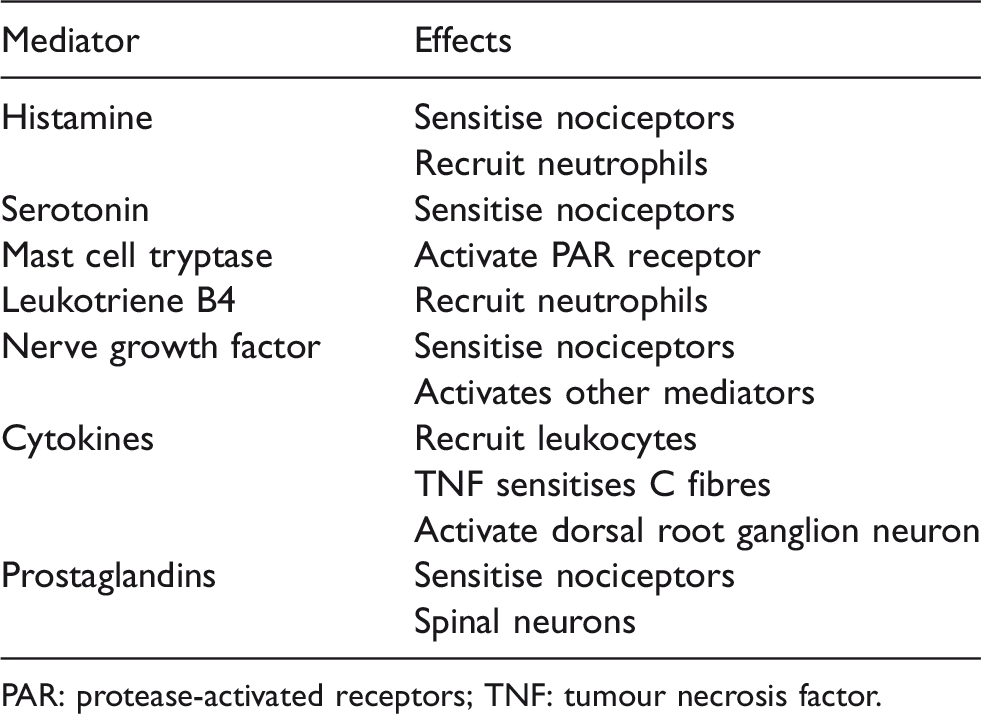
PAR: protease-activated receptors; TNF: tumour necrosis factor.
Tryptase is also released by mast cell degranulation and contributes to nociception and hyperalgesia in a rat model. 7 In a rat model of PSL, sodium cromoglycate, a mast cell stabiliser, was found to reduce neuronal hypersensitivity by inhibiting recruitment of immune cells. 8
TNF released by mast cells following peripheral nerve injury causes pain. Anti-TNF drugs such as infliximab and etanercept have been reported to be effective in treating pain associated with inflammatory conditions such as Crohn’s disease and rheumatoid arthritis9,10 (Level 3 evidence). Further evidence of the role of TNF in pain syndromes is provided by case reports of the beneficial effects of infliximab in complex regional pain syndrome 11 (Level 3 evidence). However, infliximab did not show any benefit in the treatment of sciatica in a randomised controlled trial. 12
Neutrophils
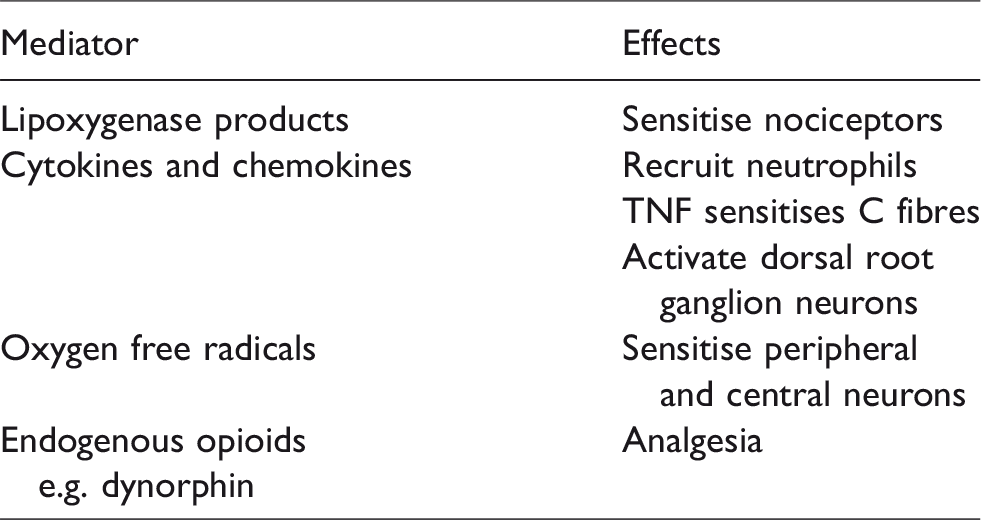
Although normally not present in intact nerves, neutrophils are the earliest inflammatory cells that infiltrate damaged nerves and tissues. 5 They are recruited to the damaged nerve by chemotactic factors, including NGF-β, CXCL1, monocyte chemoattractant protein-1 (MCP-1), leukotriene-B4 and bacterial formyl peptides. Endoneurial neutrophil infiltration into the damaged nerve usually peaks within the first three days after injury. 13 Neutrophils initiate phagocytosis, release pro-inflammatory mediators and recruit other immune cells (e.g. mast cells and macrophages). Hyperalgesia occurs as a result of the release of pro-inflammatory mediators from the neutrophils. These include prostacyclin, bacterial proteins, proteases, 155-dihydroxyeicosatetraenoic acid (15S-diHETE), TNF-α, IL-1β, IL-2, IL-6, bradykinin, hydrogen ions, superoxide and reactive oxygen species (ROS) 2 (Table 5). Neutralising antibodies against neutrophils administered within the first 48 hours after injury decreases allodynia in a rat model. 14 However, it is not effective after this period.
Neuroimmune mediators of neutrophils.
Macrophages
Macrophages remove microbes, leukocytes and damaged cells by phagocytosis. Following nerve injury, resident macrophages proliferate. Circulating monocytes are also recruited to the site of injury. This process is mediated by chemokines such as MCP-1, macrophage inflammatory protein-1α, IL-1β and defensin. 15 These macrophages infiltrate damaged nerves and the dorsal root ganglia. This process peaks at 3–13 days after injury. 13 A further release of pro-inflammatory mediators, such as TNF, IL-1β, IL-6, cyclooxygenase-2 (COX-2), cathepsin S, ROS and prostaglandins, sensitise primary afferent nerves, resulting in hyperalgesia 5 (Table 6).
Neuroimmune mediators of macrophages.
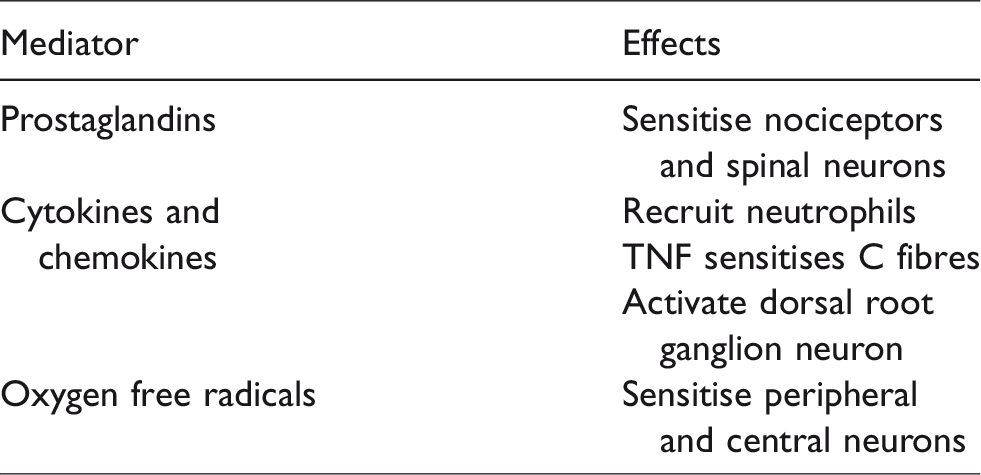
When an axon is transected, Wallerian degeneration occurs. The distal axons degenerate, and the Schwann cell de-differentiate. 15 Macrophages accumulate in degenerating nerves and clear the debris. Inflammatory cells are recruited and cytokines are released, and these cause pain hypersensitivity. Studies using human nerve biopsies have demonstrated that the magnitude of cytokine response was correlated with the severity of neuropathic pain. 16 Prostaglandins released by macrophages directly sensitise primary afferent nerves and are also implicated in neuropathic pain. This is supported by the findings of Syriatowicz et al. 17 who demonstrated that COX inhibitors suppressed macrophage-induced prostaglandin release and reduced hyperalgesia in rat models of PSL.
Cathepsin S, a lysosomal cysteine protease which is expressed by macrophages infiltrating the dorsal root ganglia and site of nerve injury, is up-regulated in rat dorsal ganglia following peripheral nerve injury. Barclay et al. 18 found that cathepsin S inhibition reversed mechanical hyperalgesia but not allodynia. They suggested that endogenous cathepsin S released by macrophages sustained hyperalgesia following nerve injury.
Additional evidence of the role of macrophages in the pathogenesis of neuropathic pain is supported by studies using drugs that deplete macrophages. Liu et al. 19 demonstrated that clodronate, a macrophage depleting agent, decreased thermal hyperalgesia and nerve fibre degeneration in PSL models of neuropathic pain. However, Rutkowski et al. reported that clodronate and CNI-1493 had only limited analgesic effects in PSL. 20 Overall, the exact role of drugs targeting macrophages remains unclear.
Glial cells
Glial cells are non-neuronal cells that surround and support neurons. These include Schwann and satellite cells in the peripheral nervous system and microglia and astrocytes in the CNS. Microglia and astrocytes are found in supraspinal centres (the rostral ventromedial medulla, hypothalamus and periaqueductal grey area) and are involved in the descending pain pathways. 13 At rest, glial cells provide structural integrity, modulate neurotransmission, secrete neurotrophic factors, destroy and clear cellular pathogens and debris, and do not have a role in pain modulation. 21
An extensive immune response occurs around the intact as well as injured cell bodies of sensory neurons following nerve injury. The proximity of glial cells to neurons that mediate pain facilitates neuroimmune interaction and maintenance of chronic and neuropathic pain.21,22 Following peripheral nerve injury, neuronal excitability is enhanced by up-regulation of pro-inflammatory mediator receptors on glial cells in the dorsal root ganglia. 22
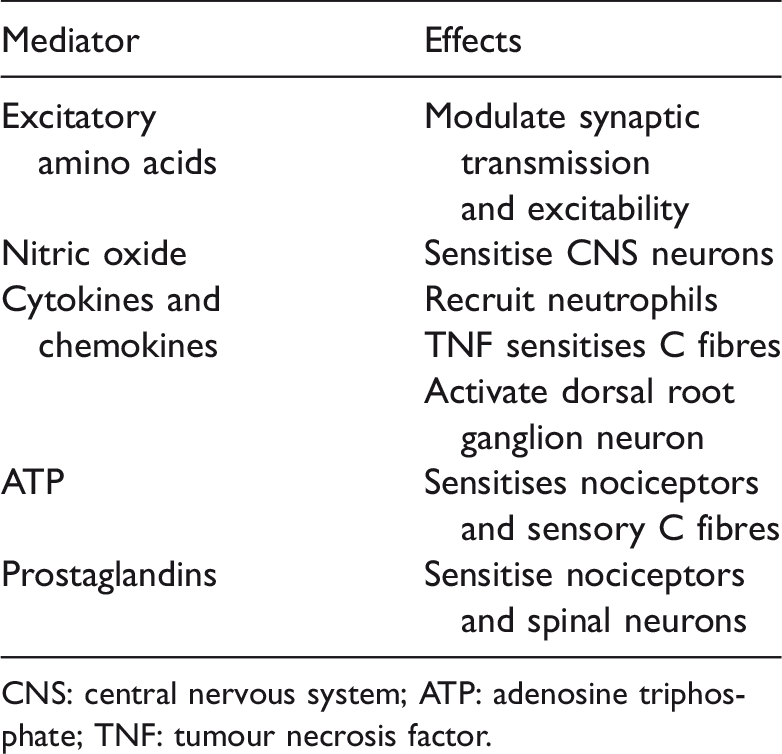
Widespread glial activation at all levels of the ascending pain pathways from the peripheral nerve to the dorsal root ganglion and higher centres 5 results from neuronal and humoral interaction (‘cross-talk’) between glial cells and neurons. 23 This ‘cross-talk’ activates a series of cascades that facilitate neuropathic pain. These cascades include mitogen-activated protein kinase (MAPK), nuclear factor kappa-light-chain enhancer of activated B-cells (NF-κB), Janus tyrosine kinase/signal transducer and activator of transcription, phosphoinositide 3-kinase/Akt kinase and Src family kinase pathways 24 (Figure 2).
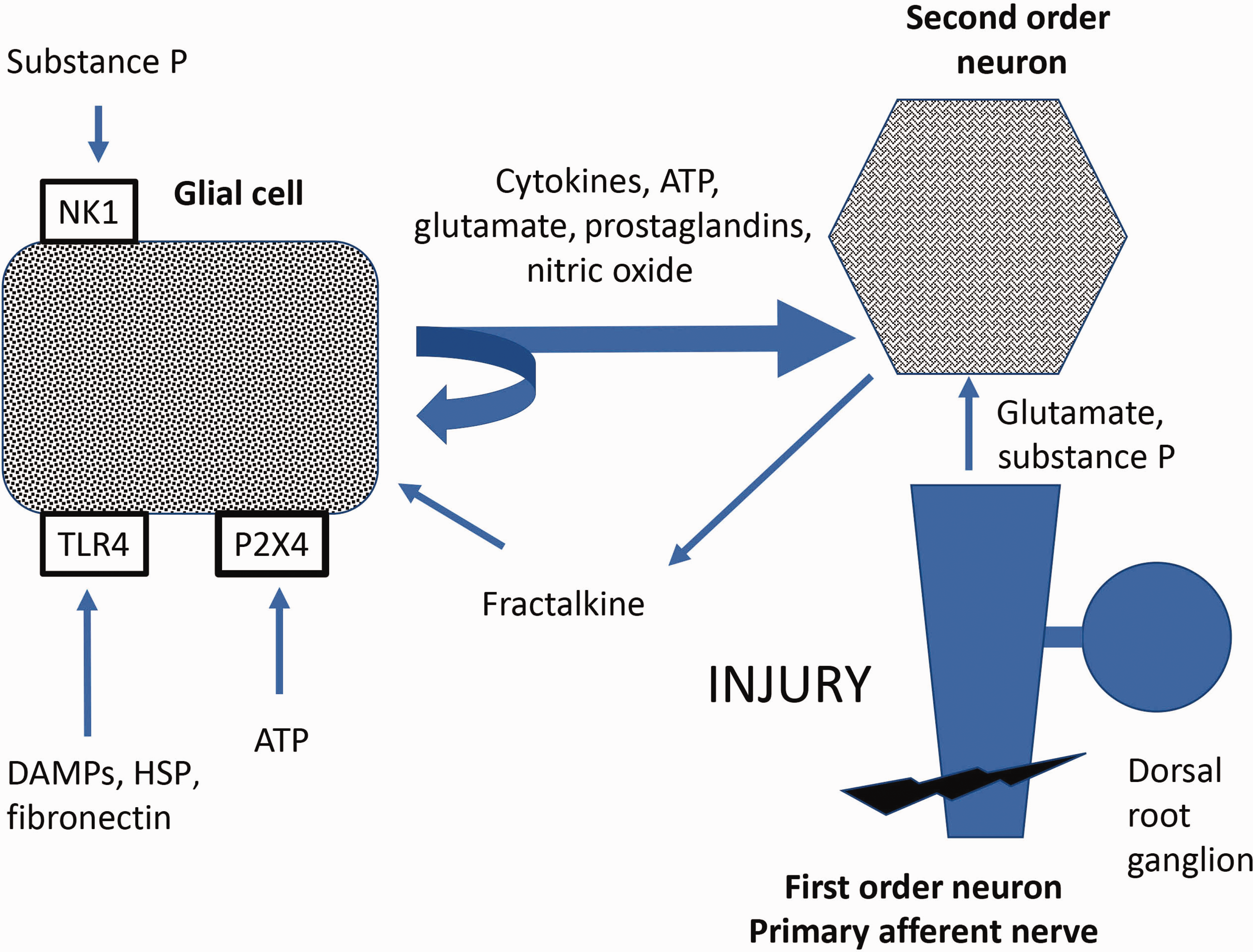
Glial activation. NK1: neurokinin 1; TLR4: Toll-like receptor subtype 4; P2X4: purinergic receptor; DAMP: danger associated molecular patterns; HSP: heat shock protein; ATP: adenosine triphosphate.
Glial activation in the dorsal horn releases substance P and glutamate that stimulate primary afferent and second-order neurons. 21 Sustained depolarisation of the second-order neurons activates N-methyl-D-aspartate receptors, and this results in enhanced neuronal connectivity and remodelling in the dorsal horn with subsequent central sensitisation. 25
The triggers of glial activation include trauma, infection, ischaemia, neurodegeneration, nerve injury, Wallerian degeneration and opioid administration. 22 The release of ATP, bradykinin, substance P, prostaglandins and fractalkine from surrounding sensory neurons is enhanced. 15 The number and size of activated glial cells increase, a process known as ‘reactive gliosis’. 22 Pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, prostaglandin E2 (PGE2), COX-2, nitric oxide (NO), ATP, BDNF and IL-18 during reactive gliosis 25 recruit circulating immune cells to the site of injury and pain modulation centres, and more pro-inflammatory cytokines are released. 22 Consequently, increased nociception and central sensitisation contribute to a transition of acute to chronic pain and persistent neuropathic pain. 26 Reactive gliosis is also implicated in the development of opioid-induced hyperalgesia, tolerance and dependence, as discussed later. 27
Some activated glial cells remain in a ‘primed’ state and undergo epigenetic changes that enhance their transcriptional processes. 28 Factors such as stress, ageing, illness, injury and opioid consumption increase the likelihood of immune priming. Subsequent noxious stimuli result in an exaggerated pain response as a result of increased neurotransmitter release, 26 and this contributes to the transition from acute to chronic pain. 28
Schwann cells
Schwann cells which are in close contact with the peripheral sensory nerves express major histocompatibility complex (MHC) class I molecules on their cell membranes. Bergsteinsdottir et al. 29 showed that expression of MHC class II and MHC class I molecules was up-regulated when rat Schwann cells were cultured with T lymphocytes and stimulated with interferon-γ. These activated Schwann cells 5 enhanced the release of inflammatory cytokines such as TNF, IL-1, IL-6, NGF, PGE2, ATP, leukaemia inhibitory factor and MCP-1 (Table 7). Wallerian degeneration following nerve transection leads to phagocytosis of myelin debris by Schwann cells, and algesic mediators such as cytokines (TNF and interleukins), ATP and nerve growth factor are released. Using a chronic constriction injury (CCI) rat model, Campana et al. 30 demonstrated that reduced TNF immunoreactivity in Schwann cells was associated with antinociceptive and antineuropathic effects of recombinant erythropoietin.
Neuroimmune mediators of glial and Schwann cells.
CNS: central nervous system; ATP: adenosine triphosphate; TNF: tumour necrosis factor.
Satellite glial cells
Satellite glial cells are the peripheral cells that surround the nerve cell bodies within the dorsal root ganglia. They regulate the dorsal root ganglia and facilitate sensory transmission and nociceptive pathways. At rest, satellite glial cells buffer glutamate and release neurotrophic factors which protect the nerve cells. 5 Following peripheral nerve injury, the satellite cells proliferate and release ATP, TNF, IL-1β and NGF-β. 31 Animal studies have shown that the release of such pro-inflammatory mediators is sustained for months after the initial injury associated with increased ectopic neuronal discharges that exacerbate pain and hyperalgesia and neuropathic pain syndromes. 5
Microglia
Microglia are resident macrophages which regulate the CNS micro-environment. When the CNS is damaged or invaded by pathogens, the microglia proliferate (‘microgliosis’). 5 Microgliosis occurs at the sensory afferent nerve terminals within the dorsal horn of the spinal cord and the ventral horn around the cell bodies of the damaged motor neurons. Microglia activated by cytokines released from neurons and glial cells become phagocytes and antigen-presenting cells. In animal models, microglia activation occurs within four hours after nerve injury and persists for several months. 5 Products of tissue injury such as ATP (acting on purinergic receptors (P2X4, P2X7 and P2Y12), nuclear factors (acting on Toll-like receptors (TLR)), complement fragments, and reactive oxygen species activate microglia. 18 ATP, MCP-1 and fractalkine attract more microglia to the site of injury as well as to the dorsal root ganglion. 32 Neuronal hyperexcitability occurs as a result of glial activation. 33 Additionally, microgliosis following peripheral injury occurs at distant structures such as the thalamus, hypothalamus, periaqueductal grey and the rostral ventromedial medulla. Cytokines released by microglia modulate central sensitisation by several mechanisms. TNF enhances excitatory currents induced by glutamate, whereas IL-1β reduces inhibitory synaptic transmission. Activated microglia phagocytose GABA-ergic inhibitory interneurons, and this contributes to neuropathic pain. 33 Activated microglia also recruit T lymphocytes which maintain neuropathic pain by secreting pro-inflammatory mediators. 15
Astrocytes
Astrocytes are the dominant cells in the CNS that regulate neurotransmitter and hydrogen ion levels in the extracellular space. There is a close relationship between astrocytes and the pre- and post-synaptic processes of nerve cells (‘tripartite synapse’). 26 Following CNS injury, astrocytes are activated by IL-1β and prostaglandins, and this can persist in the spinal cord for up to three months. 5 Chronic pain, persistent hyperalgesia and hypersensitivity are associated with hypertrophy of the astrocytes, increased production of astrocytic filaments (glial fibrillary acidic protein mRNA) and the release of pro-inflammatory mediators. 32 Activated astrocytes increase calcium release and stimulate the α2δ1 receptor of neural calcium channels. Gabapentin inhibits the α2δ1 receptor and reduces astrocyte hypersensitivity. 33 Increased glial activation has been demonstrated in the spinal cords of patients with human immunodeficiency virus–related chronic pain or complex regional pain syndrome.34,35
Glial inhibitors can prevent peripheral and central sensitisation. Ibudilast, a central glial inhibitor, inhibits mechanical allodynia in animal models. 36 Propentofylline, another glial inhibitor, inhibits the release of pro-inflammatory mediators and thereby reduces neuropathic pain in animal models of pain. 37 Pre-emptive use of minocycline, an inhibitor of glial activation, reduced the expression of glial markers and pain hypersensitivity in animal models of pain. 32 Minocycline suppressed glial priming in an animal study using a ‘sham’ laparotomy model. 38 The literature on the efficacy of minocycline in humans is mixed. Sumracki et al. 39 reported that minocycline decreased hyperalgesia in patients with sciatica (Level 3 evidence). However, Martinez et al. could not demonstrate any beneficial effects of minocycline in patients with lumbar radiculopathy. 40 Inhibition of glial metabolism with methotrexate and fluorocitrate also reduced microglial activation and neuropathic pain behaviour in rat models of peripheral nerve injury. 41
Over-expression or stimulation of microglial cannabinoid CBR2 receptors reduces pain hypersensitivity in rat models of pain. 5 Austin and Moalem-Taylor 5 proposed that inhibition of spinal microglia by CBR2 agonists may offer a novel method of suppressing spinal microglia in patients with neuropathic pain.
TLRs
TLRs are transmembrane protein receptors present in dendritic cells, macrophages and glial cells.27,42 TLRs found on the Schwann cells surrounding the C fibres of primary sensory neurons, have an important role in opioid tolerance and in the initiation of both inflammation and neuropathic pain. 3 Of the 10 subtypes of TLRs in humans, the TLR2, TLR3 and TLR4 subtypes are crucial for glial activation and response in neuropathic and chronic pain mechanisms. 3
TLRs are activated by exogenous pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs). 27 PAMPs include lipopolysaccharide, flagellin, lipoproteins and RNA strands, whereas DAMPs include high mobility group box 1 protein (HMGB-1), heat shock proteins, microRNA, mitochondrial RNA, DNA strands and histones. Cell stress and injury release DAMPs such as hyaluronic acid, fibrinogen, fibronectin and biglycan. 3 Adaptor proteins (MyD88), Toll receptor associated molecule and sterile-alpha and armadillo motif containing protein are activated when TLRs bind to specific PAMPs or DAMPs and increase the release of glial pro-inflammatory mediators. 43 TLR activation releases pro-inflammatory mediators such as MCP-1, ROS, NO, IL-6, IL-1α, TNF, IL-5, IL-3, IFN β, CXCL10, CCL5, inducible NO synthase, PGE2 and calcitonin gene-related peptide. 42 TLRs mainly utilise the MyD88 pathway in the dorsal root ganglion and the C and A fibre neurons 44 (Figure 3). Opioids such as morphine specifically activate TLR4 signalling that leads to opioid tolerance and hyperalgesia. 42 In addition, activation of TLR3, TLR7 and TLR9 increases the expression and sensitivity of transient receptor potential vanilloid receptor 1 receptors. 42
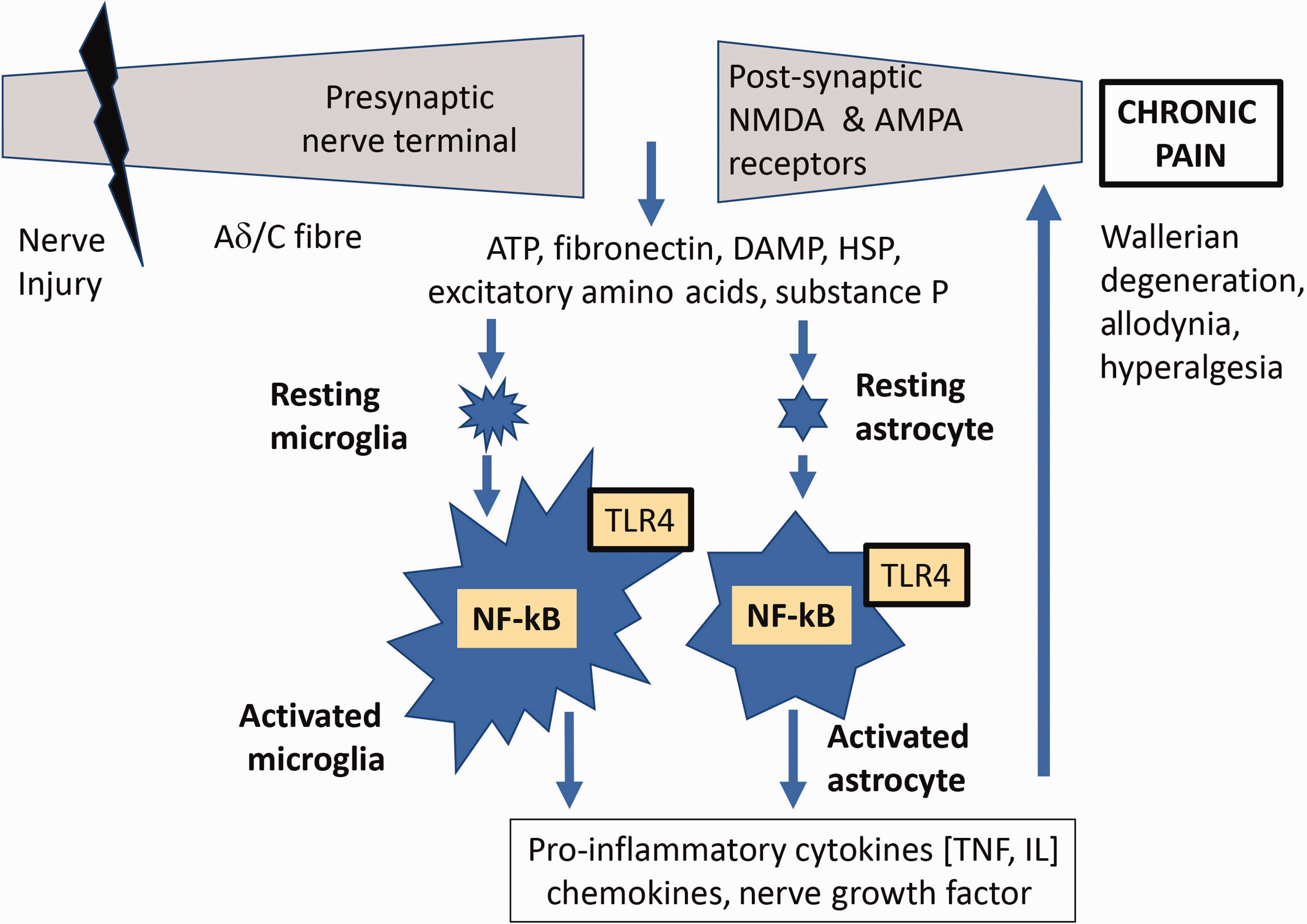
Activation of toll receptors. NMDA: N-methyl-D-aspartate; NF-κB: kappa-light-chain enhancer of activated B cells; AMPA: α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; TNF, tumour necrosis factor; IL: interleukins; ATP, adenosine triphosphate; HSP: heat shock protein; TLR4: Toll-like receptor subtype 4.
Antagonists of TLRs have analgesic properties. FP-1, a TLR4 antagonist, decreased thermal hyperalgesia and mechanical allodynia in a CCI model. 45 Rifampicin, a TLR4 and NF-κB inhibitor, decreased glial activation and allodynia in a sciatic nerve CCI model. 43 Intrathecal administration of antibodies against HMGB-1 (an endogenous TLR ligand) inhibits TLR activation and decreases allodynia in animal models of bone cancer and diabetic neuropathy. 46
A double-blind randomised controlled trial of chaperonins 10, a TLR4 antagonist, in patients with rheumatoid arthritis demonstrated that it had anti-inflammatory properties 47 (Level 2 evidence). Ibudilast, a TLR4 inhibitor, is effective as an analgesic in patients with painful diabetic neuropathy and complex regional pain syndrome28,48 (Level 3 evidence).
Opioids bind to TLRs and activate microglia. It is proposed that microglial activation may be responsible for the hyperalgesic effects of opioids 49 (Figure 4).
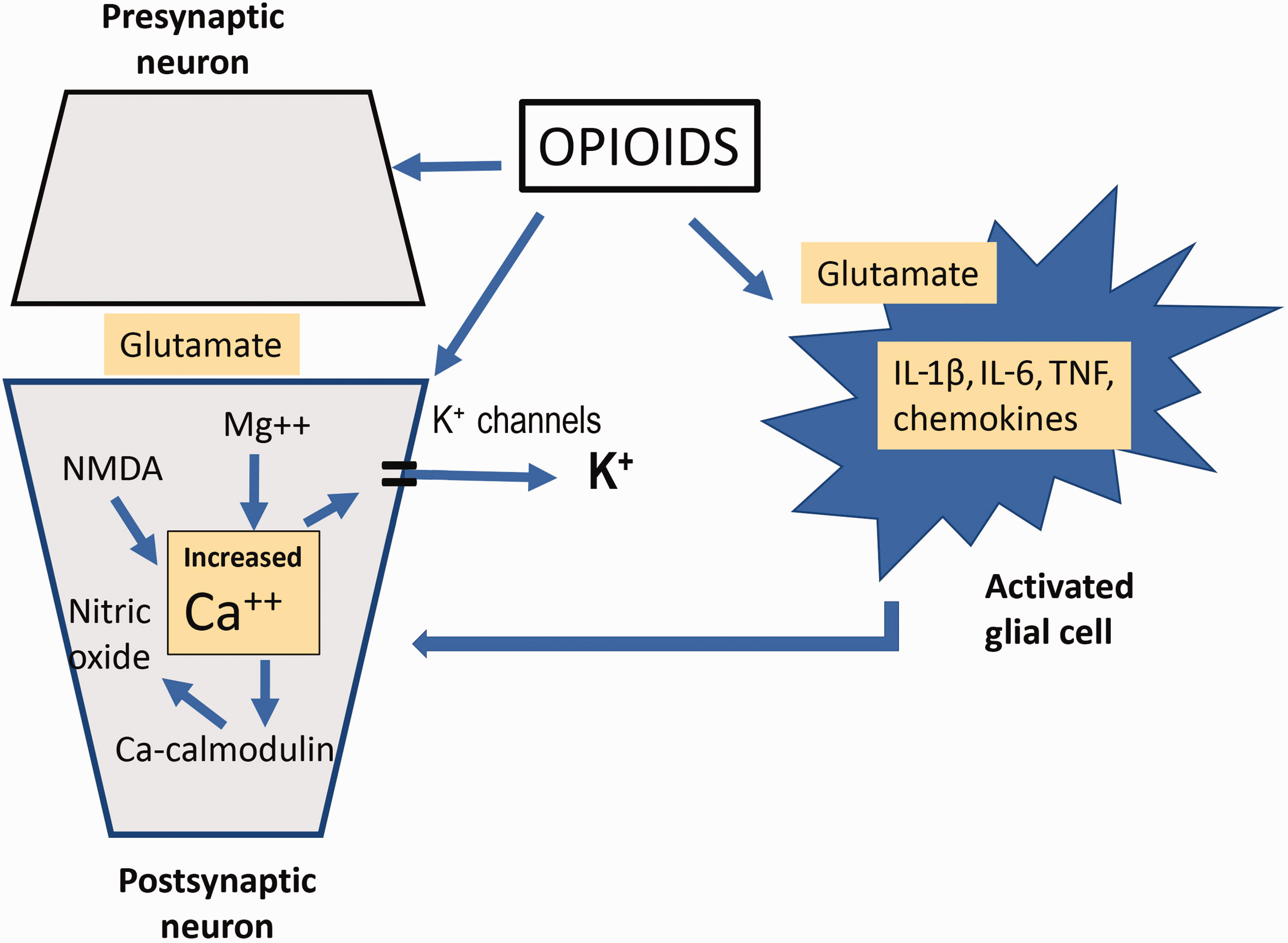
Cellular mechanisms of morphine tolerance/hyperalgesia.NMDA: N-methyl-D-aspartate; IL: interleukins; TNF: tumour necrosis factor.
Antineuropathic effects of ketamine are partly mediated by TLR3 inhibitors. 43 The antineuropathic actions of amitriptyline are partly due to its TLR4 inhibition 28 in addition to its antiserotonergic and antihistaminic properties.
T lymphocytes
T lymphocytes have a central role in adaptive immunity. 2 Helper T lymphocytes (CD4+) infiltrate the damaged nerves at about three days following injury as a result of mediators released from mast cells, macrophages and neutrophils. 21 This process peaks at approximately 21 days after injury. T lymphocytes are also recruited into injured nerves by activated microglia via MHC molecular expression. 21 Retrograde signals from the peripheral nerves increase T lymphocyte levels in the CNS and dorsal root ganglion. 21 This persists for up to 11 weeks after injury and results in persistent neuropathic and chronic pain.
Mechanical and thermal hypersensitivity following CCI is reduced in athymic mice lacking T lymphocytes. 50 An intraperitoneal injection of type 1 helper T lymphocytes reverses the reduced thermal hypersensitivity. Inhibition of T lymphocytes by leflunomide and methotrexate reduces allodynia in rat models of mononeuropathy and lumbar radiculopathy.51,52 These experimental data from rodent studies provide evidence that T lymphocyte modulation may be useful in the management of neuropathic pain.
Modulators of neuroimmune pathways
There is great interest in developing pharmacological strategies targeting the neuroimmune interface for chronic management and prevention (Table 8). These new strategies include (a) specific cytokine antagonists, (b) inhibition of pro-inflammatory signalling and (c) stimulation of anti-inflammatory signalling at the neuroimmune interface. A brief review of these investigational drugs is provided below.
Modulators of neuroimmune cells.
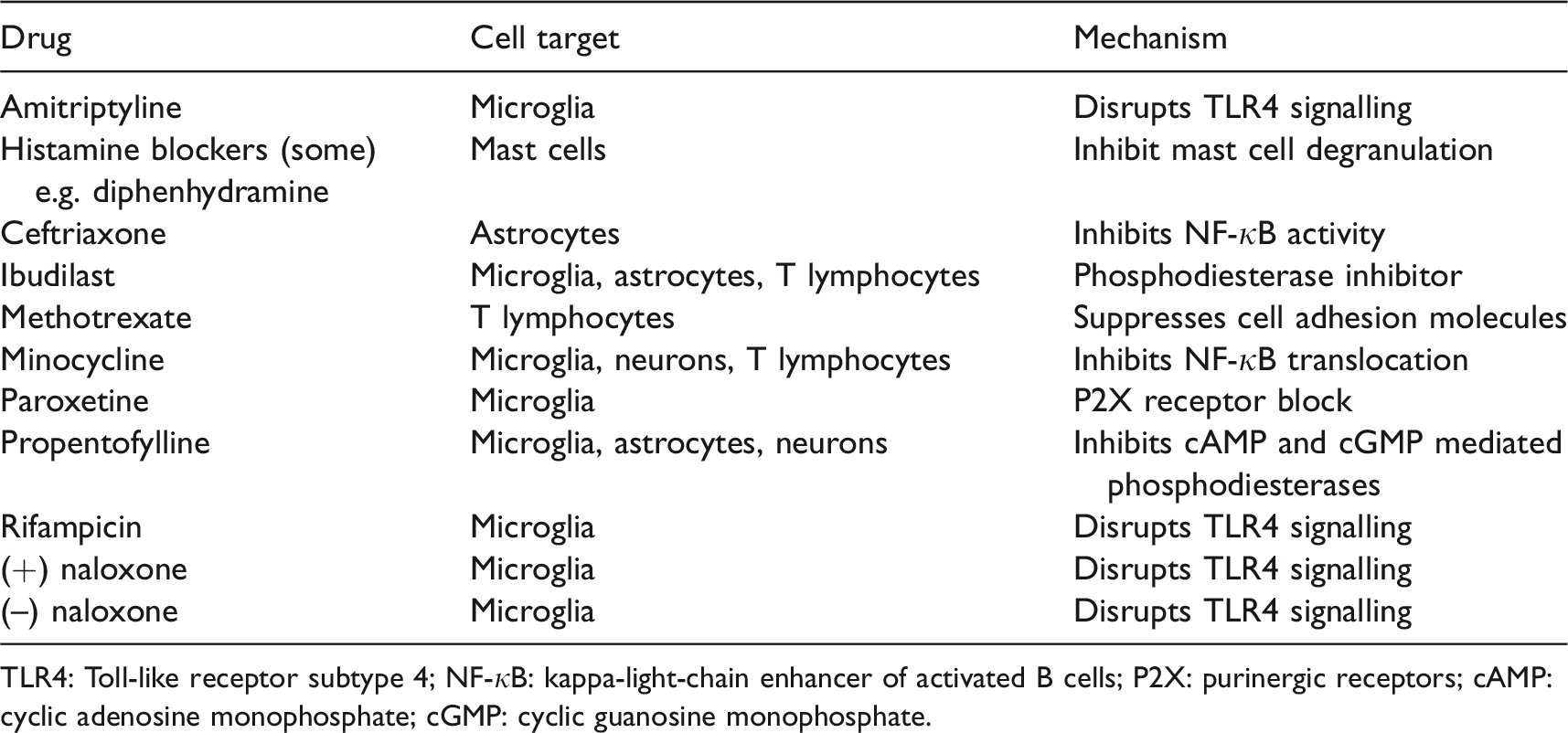
TLR4: Toll-like receptor subtype 4; NF-κB: kappa-light-chain enhancer of activated B cells; P2X: purinergic receptors; cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate.
Cytokine antagonists
Preclinical studies have shown that etanercept (TNF blocker) and IL-1Ra attenuated nociceptive hypersensitivity.53,54 However, randomised controlled trials in humans using anti-TNF-α therapy have not shown any positive results.12,55 The inability of these drugs (large molecules) to diffuse across the blood–brain barrier and the redundancy in cytokine signalling pathways may explain the lack of efficacy of these drugs.
Inhibition of pro-inflammatory signalling
Preclinical studies of minocycline, propentofylline, ibudilast and methotrexate have reported mixed clinical efficacy. The anti-inflammatory actions of minocycline are mediated by inhibition of NF-κB translocation to the nucleus in the microglia and neurons and suppression of nuclear factor of activated T cells (NFAT1). 28 Minocycline has been shown to be effective in neuropathic pain in animal models.56,57 Propentofylline inhibits cAMP (cyclic adenosine monophosphate), cGMP (cyclic guanosine monophosphate) phosphodiesterases and adenosine uptake in microglia, astrocytes and neurons, and attenuates neuropathic pain in a rat model. 58 However, a randomised clinical trial found that propentofylline was not effective in post-herpetic neuralgia probably because of differences between human and rodent microglia. 59 Ibudilast, a non-selective phosphodiesterase inhibitor of microglia, astrocytes and neurons, has been reported to have antineuropathic properties in multiple sclerosis. 48 TLR4 and purinergic receptor antagonists provide another therapeutic approach to neuropathic pain. Antidepressants such as amitriptyline currently used to treat neuropathic pain inhibit TLR4, 60 whereas paroxetine inhibits purinergic activity. 61 Rifampicin, a TLR4 antagonist which disrupts TLR4 signalling in microglia, has been found to be efficacious in rodent neuropathic pain models. 45 Inhibition of metabolism in glial cells by methotrexate (dihydrofolate reductase inhibitor) 62 has shown efficacy in preclinical models.
Stimulation of anti-inflammatory signalling at the neuroimmune interface
Another theoretical approach is to attenuate the pathological immune mechanisms of neuropathic pain by stimulating anti-inflammatory mechanisms to restore spinal dorsal horn homeostasis. This can be achieved with ceftriaxone which normalises excitatory amino acid transporter 2 activity in the astrocytes. 63 Anti-inflammatory cytokine gene therapy with IL-4 and IL-10 genes is effective in models of peripheral nerve and spinal cord injury. 64
Conclusion
Animal models of neuropathic pain following peripheral nerve injury provide strong evidence for the role of the neuroimmune interface in persistent pain, with the microglia and astrocytes playing an important role. However, the precise role of neuroimmune mechanisms in humans is more circumstantial. Many agents targeting the neuroimmune interface in animal models have demonstrated promising therapeutic potential, but evidence of the efficacy of these in humans is less clear. Differences in animal and human biology should be considered in the design and interpretation of trials of such agents.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.